

Lack of SPNS1 results in accumulation of lysolipids and lysosomal storage disease in mouse models
- PMID: 38451736
- PMCID: PMC11141868
- DOI: 10.1172/jci.insight.175462
Lack of SPNS1 results in accumulation of lysolipids and lysosomal storage disease in mouse models
Abstract
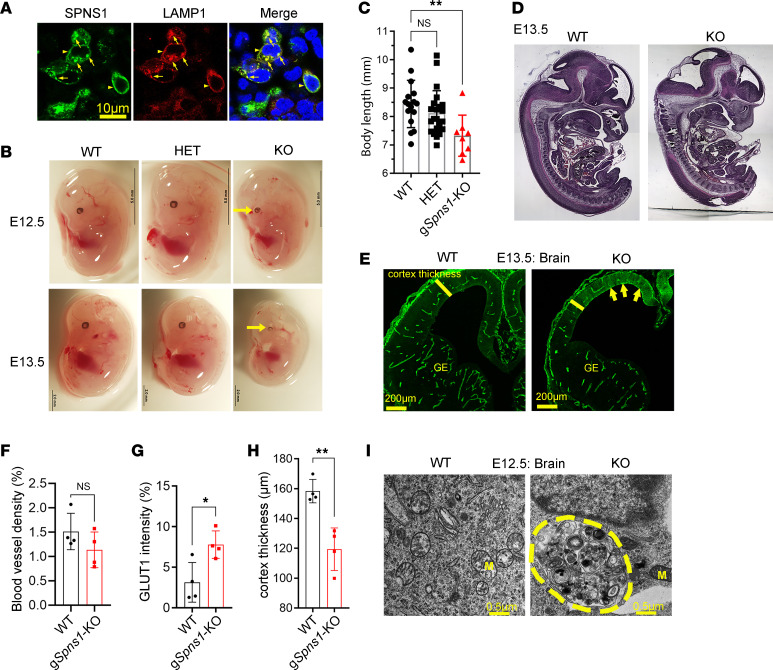
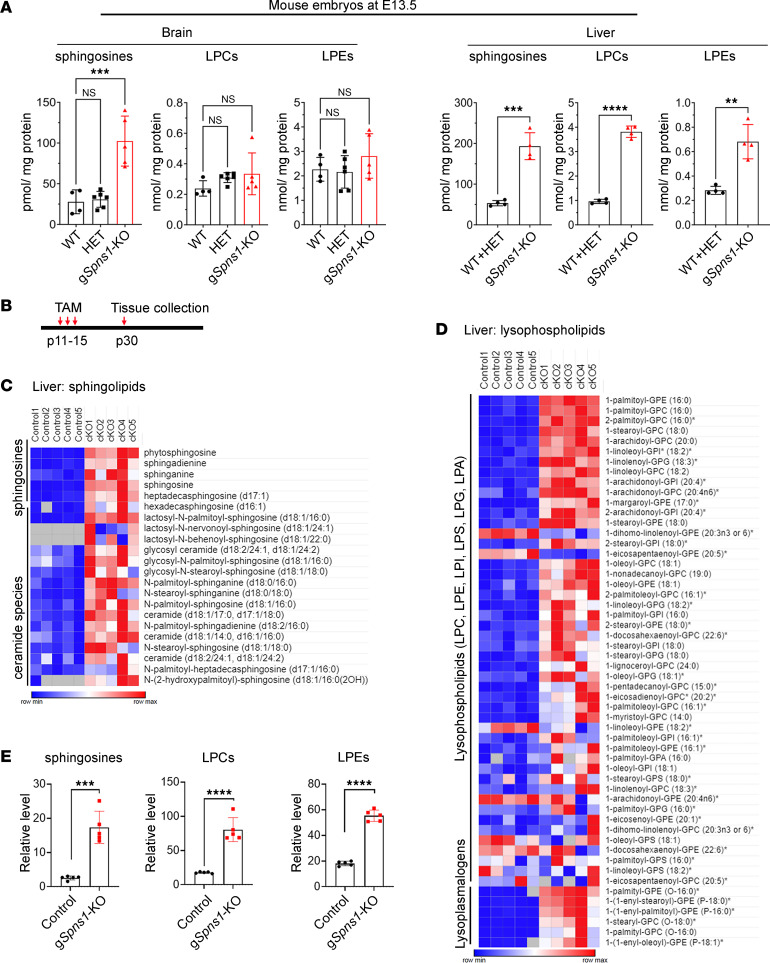
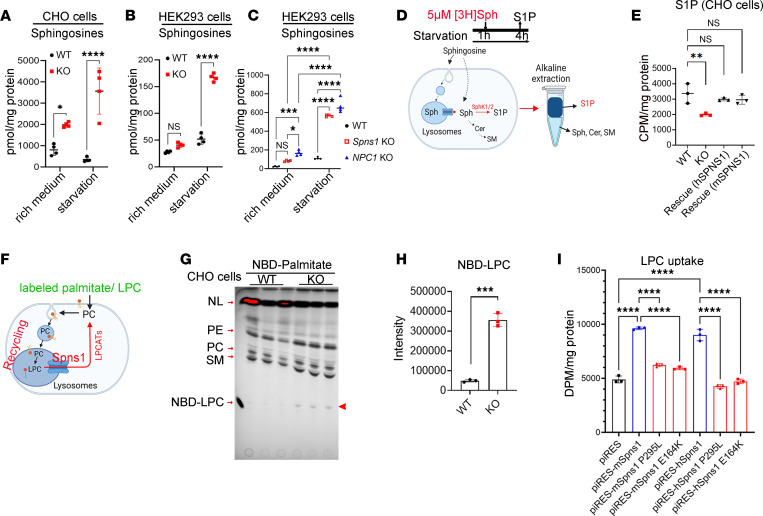
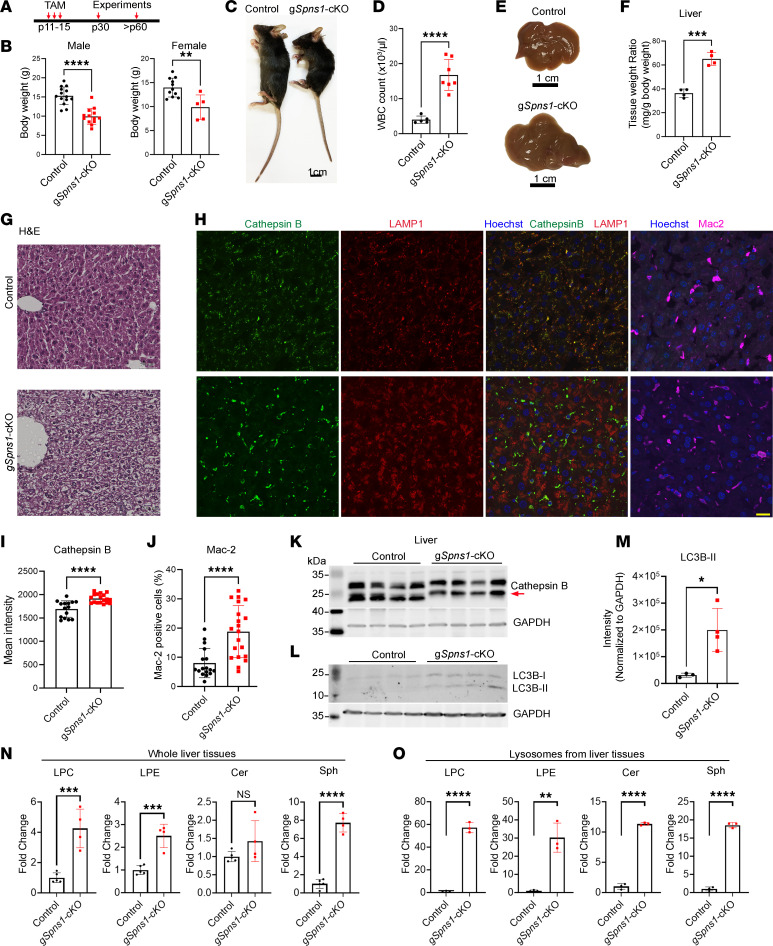
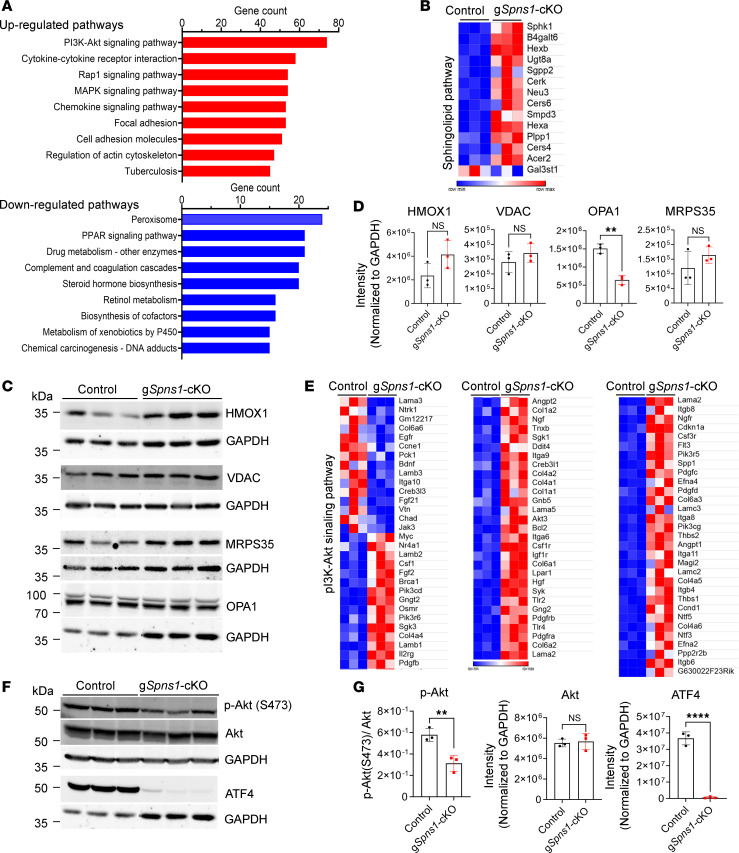
Accumulation of sphingolipids, especially sphingosines, in the lysosomes is a key driver of several lysosomal storage diseases. The transport mechanism for sphingolipids from the lysosome remains unclear. Here, we identified SPNS1, which shares the highest homology to SPNS2, a sphingosine-1-phosphate (S1P) transporter, functions as a transporter for lysolipids from the lysosome. We generated Spns1-KO cells and mice and employed lipidomic and metabolomic approaches to reveal SPNS1 ligand identity. Global KO of Spns1 caused embryonic lethality between E12.5 and E13.5 and an accumulation of sphingosine, lysophosphatidylcholines (LPC), and lysophosphatidylethanolamines (LPE) in the fetal livers. Similarly, metabolomic analysis of livers from postnatal Spns1-KO mice presented an accumulation of sphingosines and lysoglycerophospholipids including LPC and LPE. Subsequently, biochemical assays showed that SPNS1 is required for LPC and sphingosine release from lysosomes. The accumulation of these lysolipids in the lysosomes of Spns1-KO mice affected liver functions and altered the PI3K/AKT signaling pathway. Furthermore, we identified 3 human siblings with a homozygous variant in the SPNS1 gene. These patients suffer from developmental delay, neurological impairment, intellectual disability, and cerebellar hypoplasia. These results reveal a critical role of SPNS1 as a promiscuous lysolipid transporter in the lysosomes and link its physiological functions with lysosomal storage diseases.
Keywords: Aging; Autophagy; Embryonic development; Metabolism; Mouse models.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
