

CD38-RyR2 axis-mediated signaling impedes CD8+ T cell response to anti-PD1 therapy in cancer
- PMID: 38451948
- PMCID: PMC10945783
- DOI: 10.1073/pnas.2315989121
CD38-RyR2 axis-mediated signaling impedes CD8+ T cell response to anti-PD1 therapy in cancer
Abstract
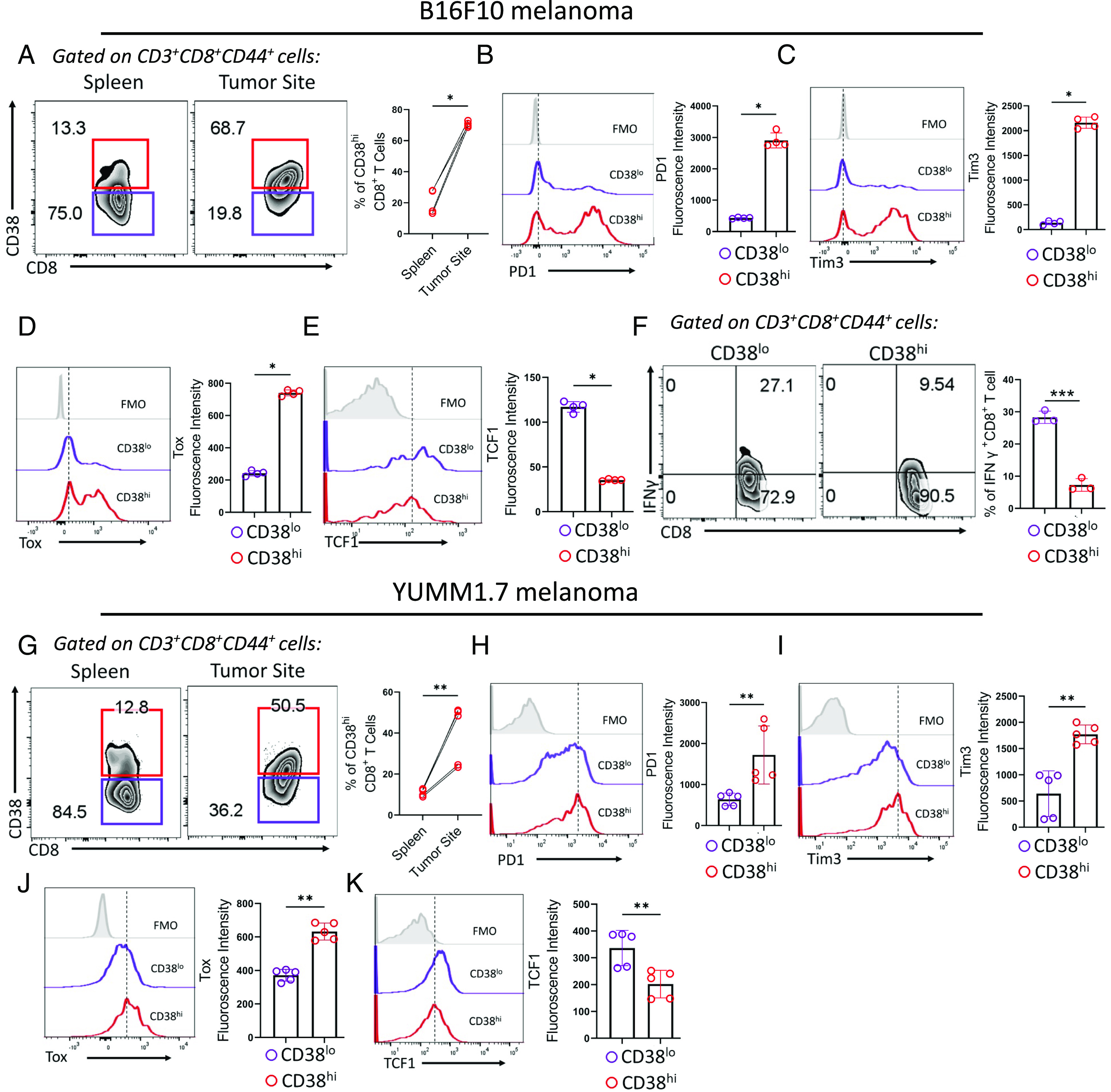
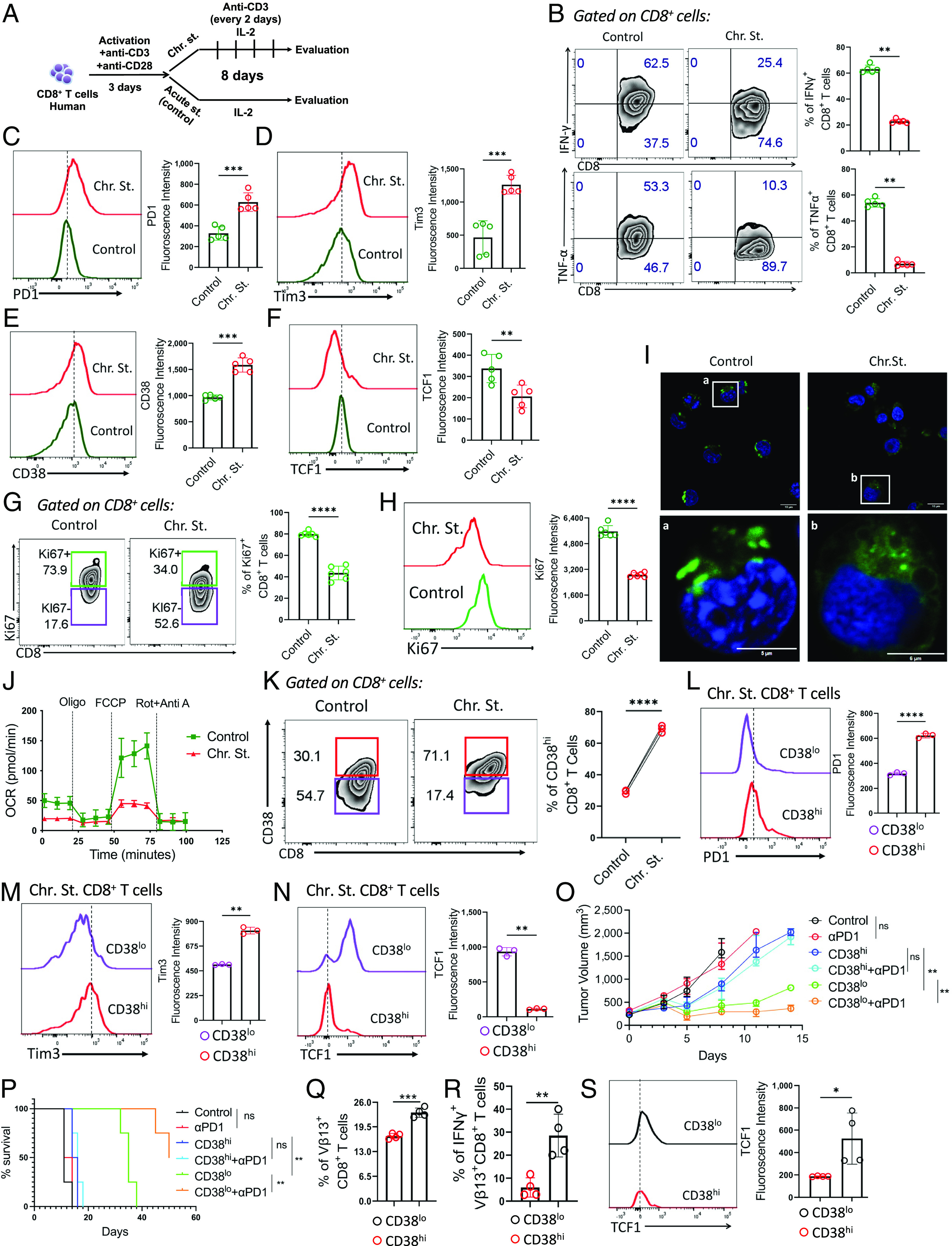
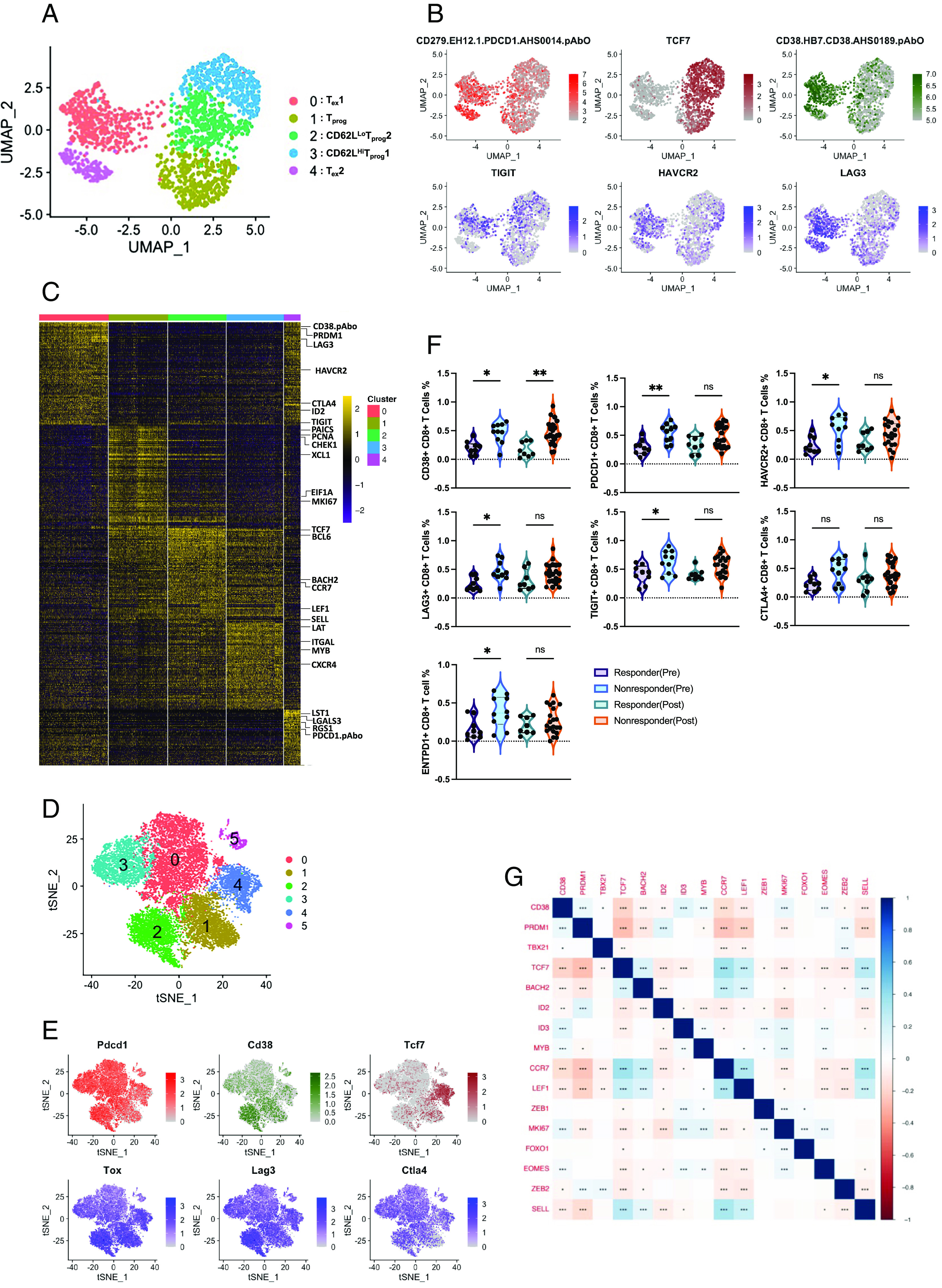
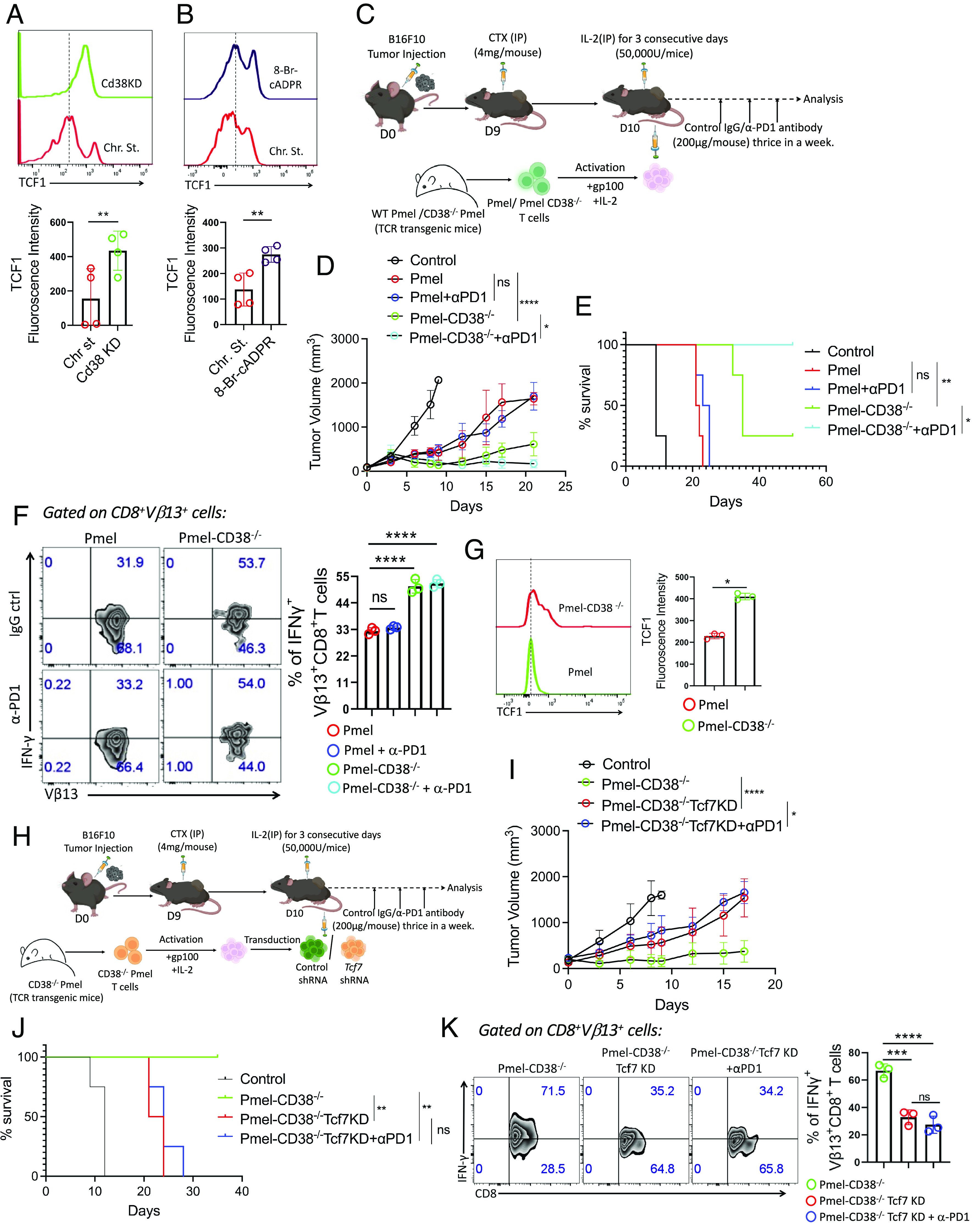
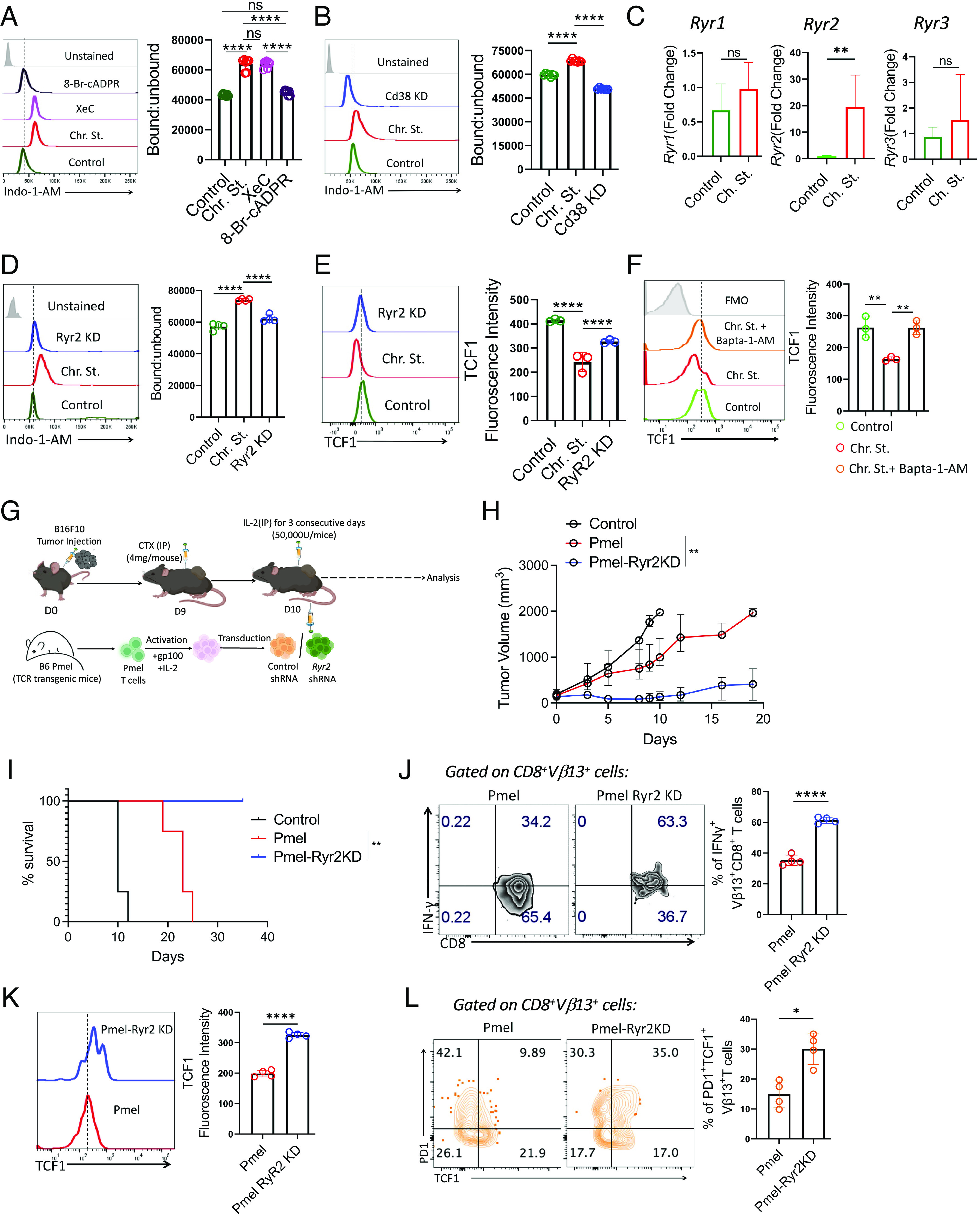
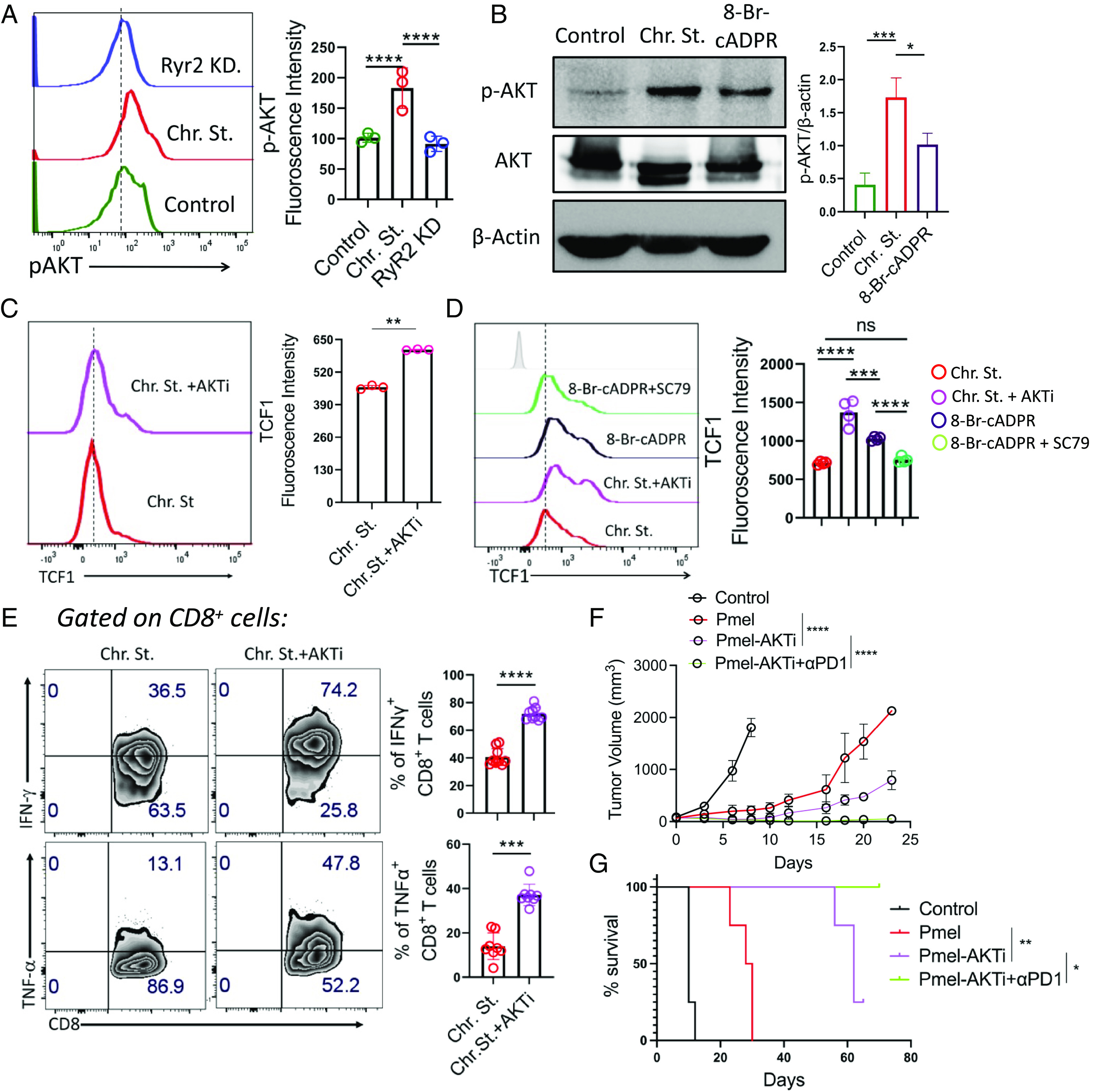
PD1 blockade therapy, harnessing the cytotoxic potential of CD8+ T cells, has yielded clinical success in treating malignancies. However, its efficacy is often limited due to the progressive differentiation of intratumoral CD8+ T cells into a hypofunctional state known as terminal exhaustion. Despite identifying CD8+ T cell subsets associated with immunotherapy resistance, the molecular pathway triggering the resistance remains elusive. Given the clear association of CD38 with CD8+ T cell subsets resistant to anti-PD1 therapy, we investigated its role in inducing resistance. Phenotypic and functional characterization, along with single-cell RNA sequencing analysis of both in vitro chronically stimulated and intratumoral CD8+ T cells, revealed that CD38-expressing CD8+ T cells are terminally exhausted. Exploring the molecular mechanism, we found that CD38 expression was crucial in promoting terminal differentiation of CD8+ T cells by suppressing TCF1 expression, thereby rendering them unresponsive to anti-PD1 therapy. Genetic ablation of CD38 in tumor-reactive CD8+ T cells restored TCF1 levels and improved the responsiveness to anti-PD1 therapy in mice. Mechanistically, CD38 expression on exhausted CD8+ T cells elevated intracellular Ca2+ levels through RyR2 calcium channel activation. This, in turn, promoted chronic AKT activation, leading to TCF1 loss. Knockdown of RyR2 or inhibition of AKT in CD8+ T cells maintained TCF1 levels, induced a sustained anti-tumor response, and enhanced responsiveness to anti-PD1 therapy. Thus, targeting CD38 represents a potential strategy to improve the efficacy of anti-PD1 treatment in cancer.
Keywords: CD38; T cell exhaustion; anti-PD1 resistance.
Conflict of interest statement
Competing interests statement:The authors declare no competing interest.
Figures
References
-
- McLane L. M., Abdel-Hakeem M. S., Wherry E. J., CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495 (2019). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
