

High niche specificity and host genetic diversity of groundwater viruses
- PMID: 38452204
- PMCID: PMC10980836
- DOI: 10.1093/ismejo/wrae035
High niche specificity and host genetic diversity of groundwater viruses
Abstract
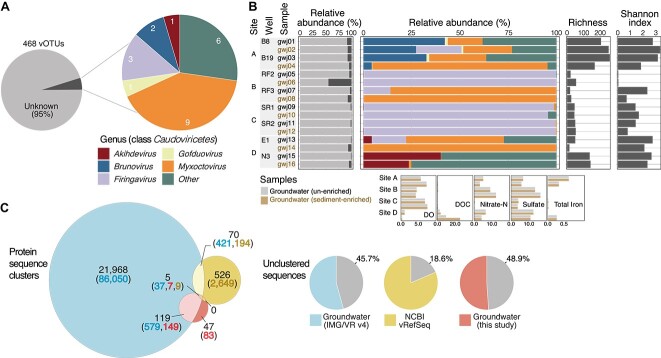
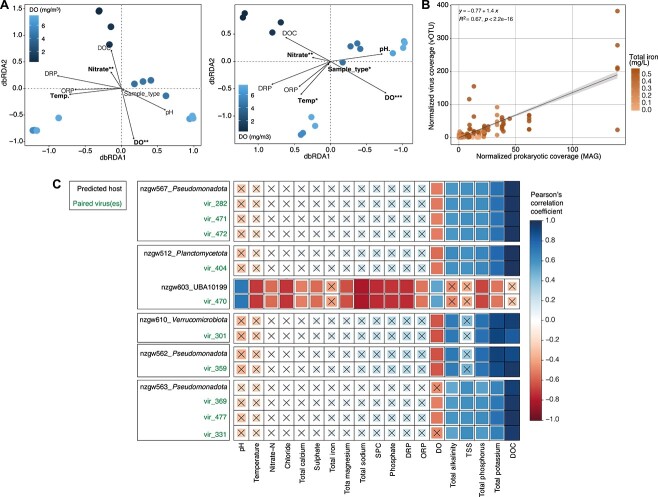
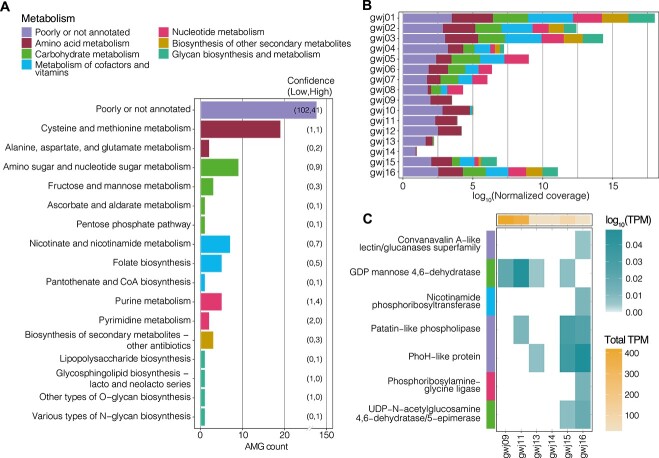
Viruses are key members of microbial communities that exert control over host abundance and metabolism, thereby influencing ecosystem processes and biogeochemical cycles. Aquifers are known to host taxonomically diverse microbial life, yet little is known about viruses infecting groundwater microbial communities. Here, we analysed 16 metagenomes from a broad range of groundwater physicochemistries. We recovered 1571 viral genomes that clustered into 468 high-quality viral operational taxonomic units. At least 15% were observed to be transcriptionally active, although lysis was likely constrained by the resource-limited groundwater environment. Most were unclassified (95%), and the remaining 5% were Caudoviricetes. Comparisons with viruses inhabiting other aquifers revealed no shared species, indicating substantial unexplored viral diversity. In silico predictions linked 22.4% of the viruses to microbial host populations, including to ultra-small prokaryotes, such as Patescibacteria and Nanoarchaeota. Many predicted hosts were associated with the biogeochemical cycling of carbon, nitrogen, and sulfur. Metabolic predictions revealed the presence of 205 putative auxiliary metabolic genes, involved in diverse processes associated with the utilization of the host's intracellular resources for biosynthesis and transformation reactions, including those involved in nucleotide sugar, glycan, cofactor, and vitamin metabolism. Viruses, prokaryotes overall, and predicted prokaryotic hosts exhibited narrow spatial distributions, and relative abundance correlations with the same groundwater parameters (e.g. dissolved oxygen, nitrate, and iron), consistent with host control over viral distributions. Results provide insights into underexplored groundwater viruses, and indicate the large extent to which viruses may manipulate microbial communities and biogeochemistry in the terrestrial subsurface.
Keywords: aquifer; auxiliary metabolic gene; bacteriophage; biogeochemical cycles; groundwater; virus.
© The Author(s) 2024. Published by Oxford University Press on behalf of the International Society for Microbial Ecology.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Breitbart M, Thompson LR, Suttle CA. et al. Exploring the vast diversity of marine viruses. Oceanography 2007;20:135–9. 10.5670/oceanog.2007.58. - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
