

DNA fragility at topologically associated domain boundaries is promoted by alternative DNA secondary structure and topoisomerase II activity
- PMID: 38452213
- PMCID: PMC11040008
- DOI: 10.1093/nar/gkae164
DNA fragility at topologically associated domain boundaries is promoted by alternative DNA secondary structure and topoisomerase II activity
Abstract
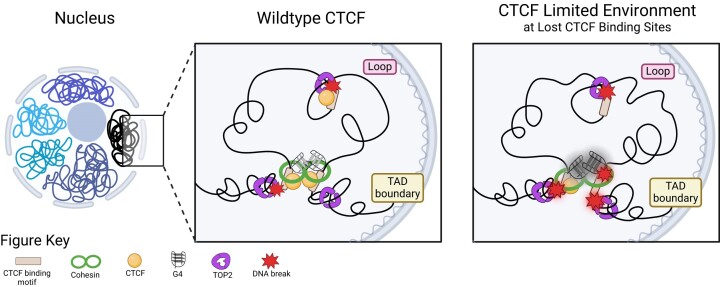
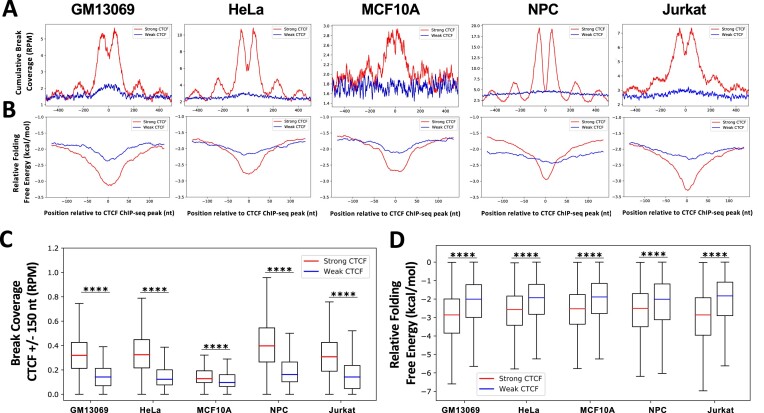
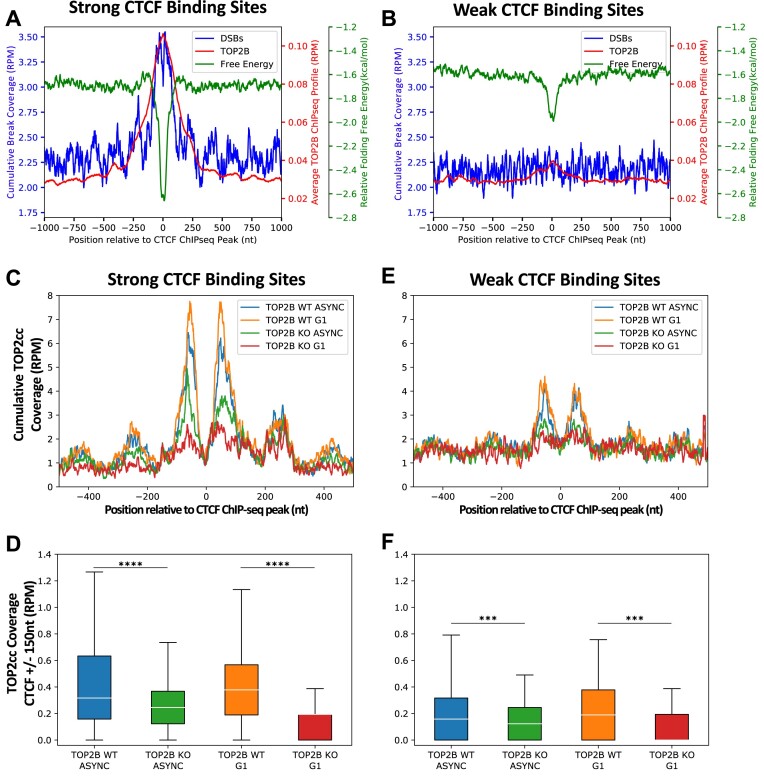
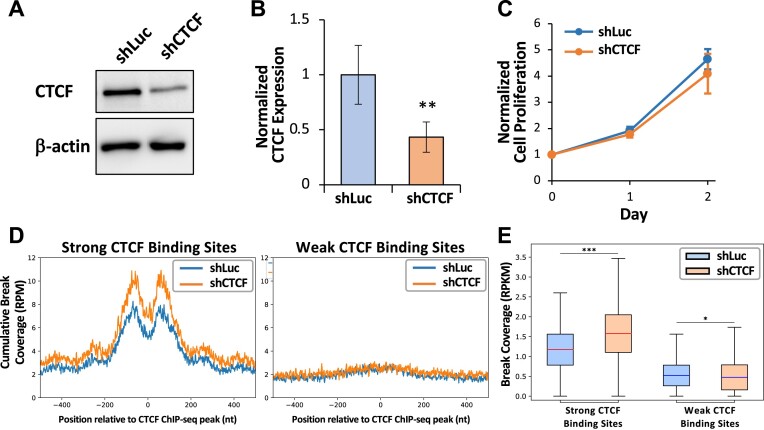
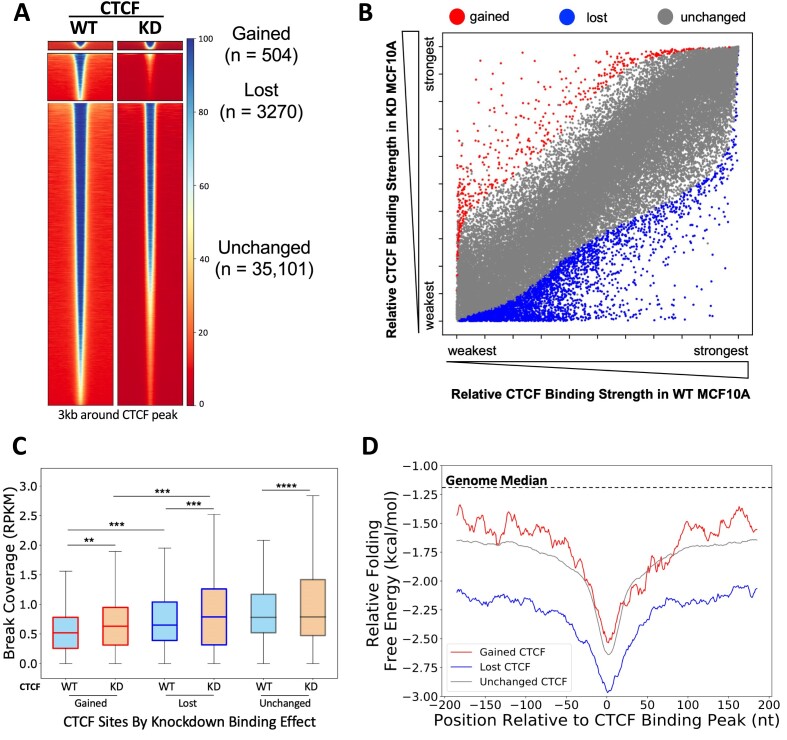
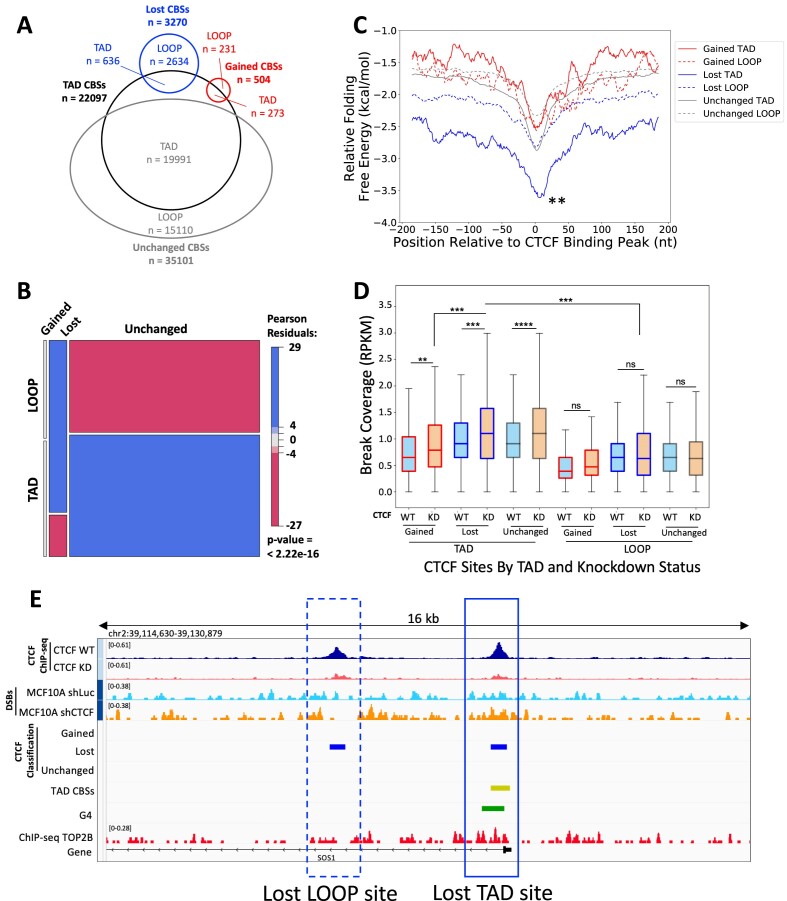
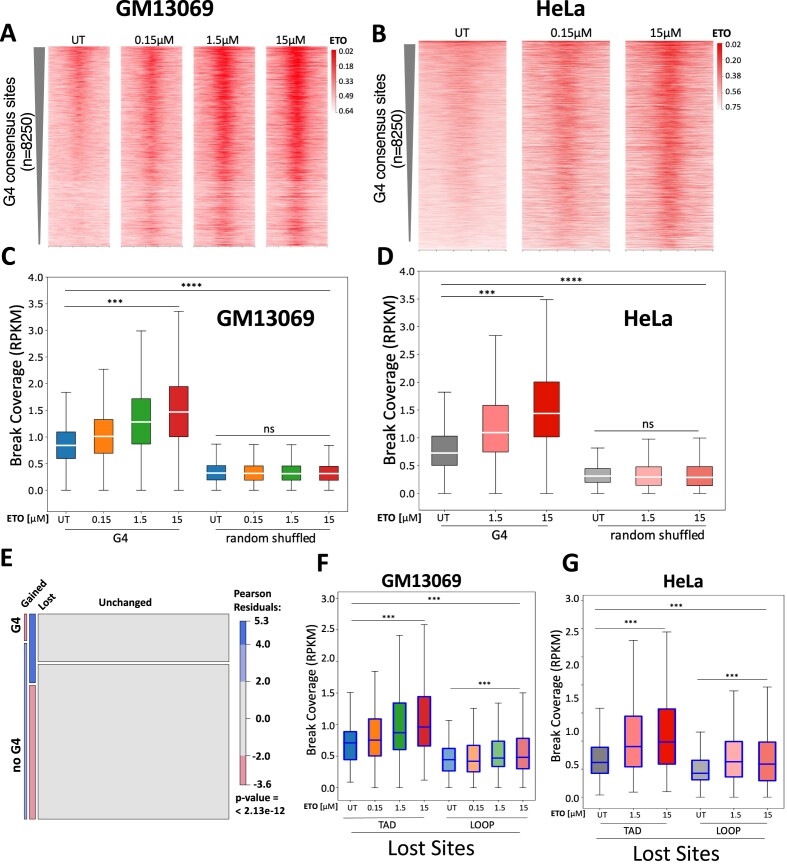
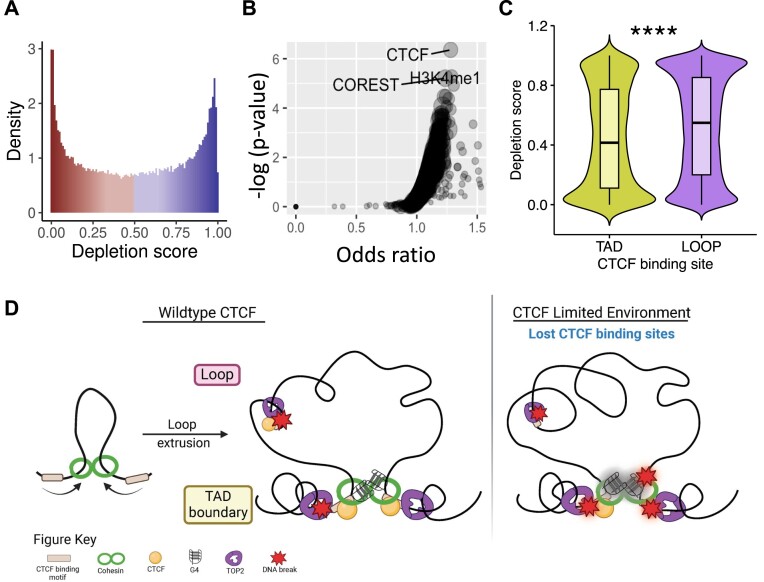
CCCTC-binding factor (CTCF) binding sites are hotspots of genome instability. Although many factors have been associated with CTCF binding site fragility, no study has integrated all fragility-related factors to understand the mechanism(s) of how they work together. Using an unbiased, genome-wide approach, we found that DNA double-strand breaks (DSBs) are enriched at strong, but not weak, CTCF binding sites in five human cell types. Energetically favorable alternative DNA secondary structures underlie strong CTCF binding sites. These structures coincided with the location of topoisomerase II (TOP2) cleavage complex, suggesting that DNA secondary structure acts as a recognition sequence for TOP2 binding and cleavage at CTCF binding sites. Furthermore, CTCF knockdown significantly increased DSBs at strong CTCF binding sites and at CTCF sites that are located at topologically associated domain (TAD) boundaries. TAD boundary-associated CTCF sites that lost CTCF upon knockdown displayed increased DSBs when compared to the gained sites, and those lost sites are overrepresented with G-quadruplexes, suggesting that the structures act as boundary insulators in the absence of CTCF, and contribute to increased DSBs. These results model how alternative DNA secondary structures facilitate recruitment of TOP2 to CTCF binding sites, providing mechanistic insight into DNA fragility at CTCF binding sites.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- O’Keefe L.V., Richards R.I. Common chromosomal fragile sites and cancer: focus on FRA16D. Cancer Lett. 2006; 232:37–47. - PubMed
-
- Ciullo M., Debily M.A., Rozier L., Autiero M., Billault A., Mayau V., El Marhomy S., Guardiola J., Bernheim A., Coullin P. et al. . Initiation of the breakage–fusion–bridge mechanism through common fragile site activation in human breast cancer cells: the model of PIP gene duplication from a break at FRA7I. Hum. Mol. Genet. 2002; 11:2887–2894. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
