

The many definitions of multiplicity of infection
- PMID: 38455332
- PMCID: PMC10910904
- DOI: 10.3389/fepid.2022.961593
The many definitions of multiplicity of infection
Abstract
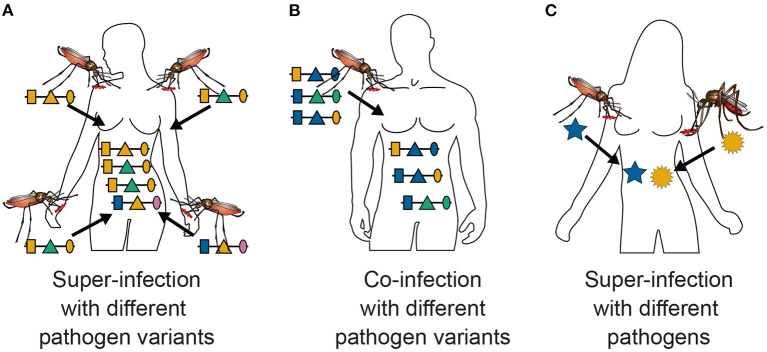
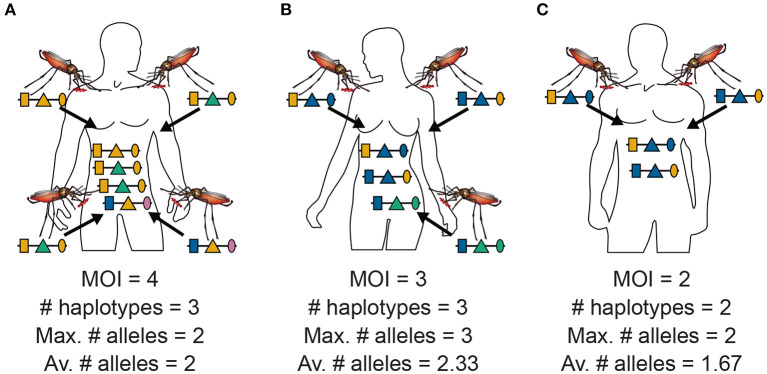
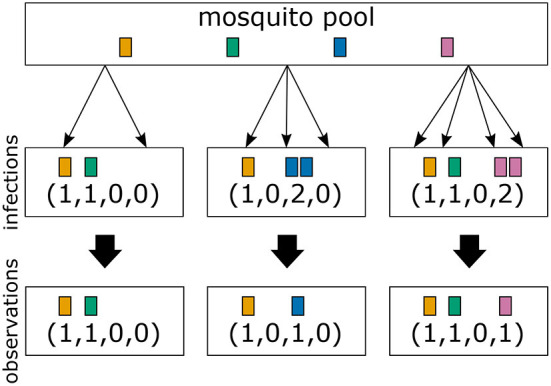
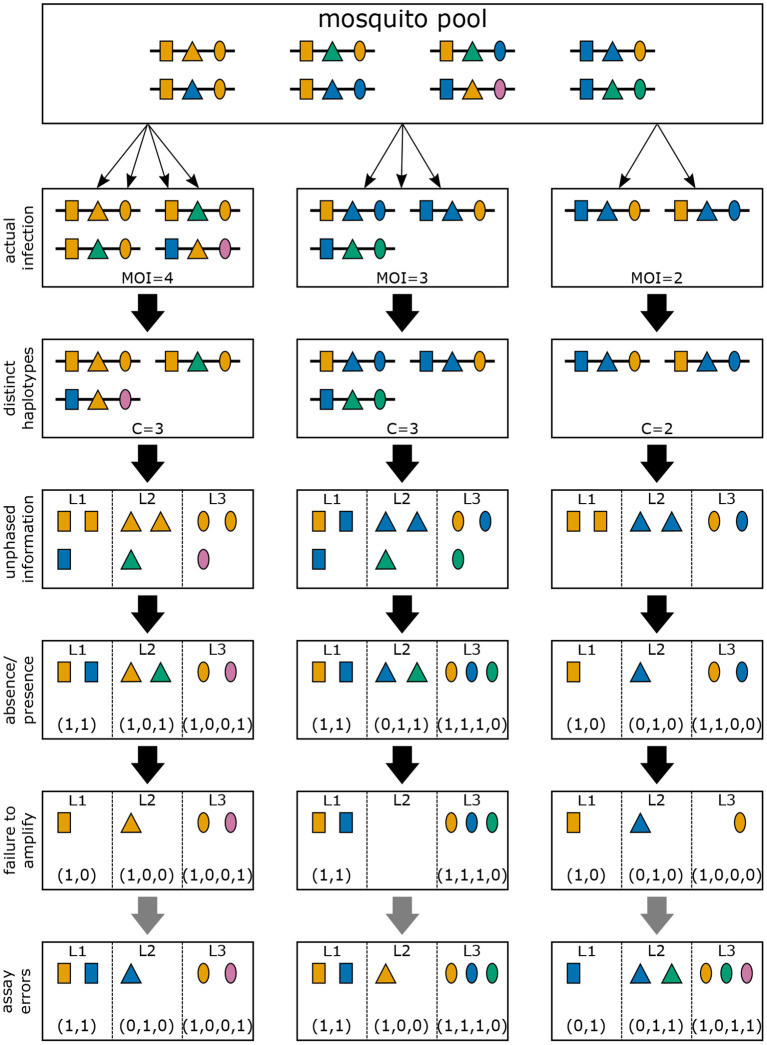
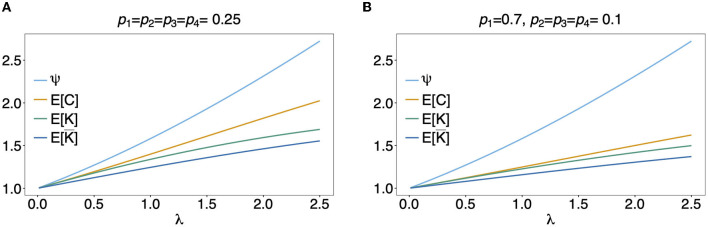
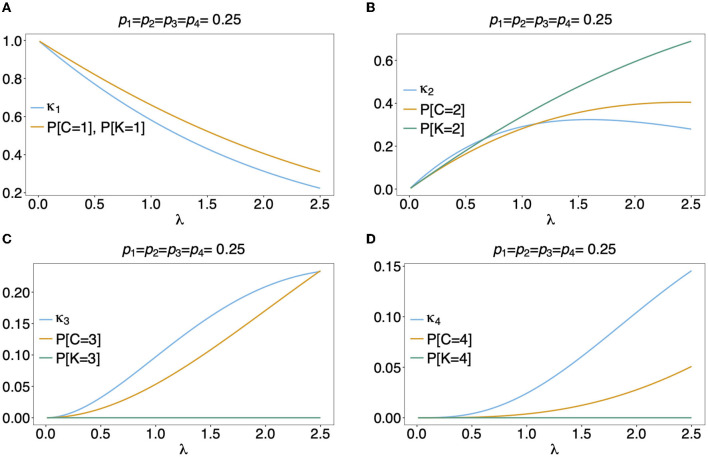
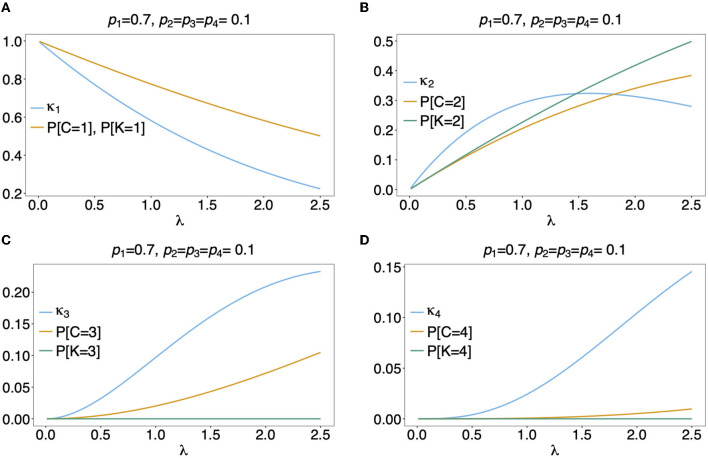
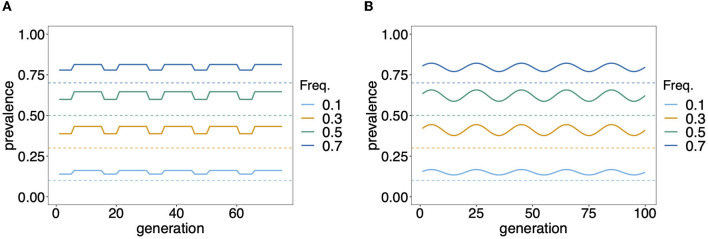
The presence of multiple genetically different pathogenic variants within the same individual host is common in infectious diseases. Although this is neglected in some diseases, it is well recognized in others like malaria, where it is typically referred to as multiplicity of infection (MOI) or complexity of infection (COI). In malaria, with the advent of molecular surveillance, data is increasingly being available with enough resolution to capture MOI and integrate it into molecular surveillance strategies. The distribution of MOI on the population level scales with transmission intensities, while MOI on the individual level is a confounding factor when monitoring haplotypes of particular interests, e.g., those associated with drug-resistance. Particularly, in high-transmission areas, MOI leads to a discrepancy between the likelihood of a haplotype being observed in an infection (prevalence) and its abundance in the pathogen population (frequency). Despite its importance, MOI is not universally defined. Competing definitions vary from verbal ones to those based on concise statistical frameworks. Heuristic approaches to MOI are popular, although they do not mine the full potential of available data and are typically biased, potentially leading to misinferences. We introduce a formal statistical framework and suggest a concise definition of MOI and its distribution on the host-population level. We show how it relates to alternative definitions such as the number of distinct haplotypes within an infection or the maximum number of alleles detectable across a set of genetic markers. It is shown how alternatives can be derived from the general framework. Different statistical methods to estimate the distribution of MOI and pathogenic variants at the population level are discussed. The estimates can be used as plug-ins to reconstruct the most probable MOI of an infection and set of infecting haplotypes in individual infections. Furthermore, the relation between prevalence of pathogenic variants and their frequency (relative abundance) in the pathogen population in the context of MOI is clarified, with particular regard to seasonality in transmission intensities. The framework introduced here helps to guide the correct interpretation of results emerging from different definitions of MOI. Especially, it excels comparisons between studies based on different analytical methods.
Keywords: MOI; co-infection; complexity of infection (COI); haplotype phasing; mixed-species infection; prevalence; super-infection; transmission intensities.
Copyright © 2022 Schneider, Tsoungui Obama, Kamanga, Kayanula and Adil Mahmoud Yousif.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Review of MrsFreqPhase methods: methods designed to estimate statistically malaria parasite multiplicity of infection, relatedness, frequency and phase.Malar J. 2024 Oct 15;23(1):308. doi: 10.1186/s12936-024-05119-2. Malar J. 2024. PMID: 39407242 Free PMC article. Review.
-
Estimating multiplicity of infection, haplotype frequencies, and linkage disequilibria from multi-allelic markers for molecular disease surveillance.PLoS One. 2025 May 27;20(5):e0321723. doi: 10.1371/journal.pone.0321723. eCollection 2025. PLoS One. 2025. PMID: 40424286 Free PMC article.
-
Estimating multiplicity of infection, allele frequencies, and prevalences accounting for incomplete data.PLoS One. 2024 Mar 21;19(3):e0287161. doi: 10.1371/journal.pone.0287161. eCollection 2024. PLoS One. 2024. PMID: 38512826 Free PMC article.
-
A maximum-likelihood method to estimate haplotype frequencies and prevalence alongside multiplicity of infection from SNP data.Front Epidemiol. 2022 Sep 23;2:943625. doi: 10.3389/fepid.2022.943625. eCollection 2022. Front Epidemiol. 2022. PMID: 38455338 Free PMC article.
-
Molecular approaches to determine the multiplicity of Plasmodium infections.Malar J. 2018 Apr 23;17(1):172. doi: 10.1186/s12936-018-2322-5. Malar J. 2018. PMID: 29685152 Free PMC article. Review.
Cited by
-
Evolutionary genetics of malaria.Front Genet. 2022 Nov 3;13:1030463. doi: 10.3389/fgene.2022.1030463. eCollection 2022. Front Genet. 2022. PMID: 36406132 Free PMC article.
-
Review of MrsFreqPhase methods: methods designed to estimate statistically malaria parasite multiplicity of infection, relatedness, frequency and phase.Malar J. 2024 Oct 15;23(1):308. doi: 10.1186/s12936-024-05119-2. Malar J. 2024. PMID: 39407242 Free PMC article. Review.
-
Soil Giant Phage: Genome and Biological Characteristics of Sinorhizobium Jumbo Phage.Int J Mol Sci. 2024 Jul 5;25(13):7388. doi: 10.3390/ijms25137388. Int J Mol Sci. 2024. PMID: 39000497 Free PMC article.
-
A Probabilistic Synthesis of Malaria Epidemiology: Exposure, Infection, Parasite Densities, and Detection.medRxiv [Preprint]. 2025 Mar 25:2025.03.24.25324561. doi: 10.1101/2025.03.24.25324561. medRxiv. 2025. PMID: 40196243 Free PMC article. Preprint.
-
Estimating multiplicity of infection, haplotype frequencies, and linkage disequilibria from multi-allelic markers for molecular disease surveillance.PLoS One. 2025 May 27;20(5):e0321723. doi: 10.1371/journal.pone.0321723. eCollection 2025. PLoS One. 2025. PMID: 40424286 Free PMC article.
References
-
- Murray CJ, Lopez, AD, Mathers, CD,. The Global Epidemiology of Infectious Diseases. Geneva: WHO (2004). Available online at: https://apps.who.int/iris/handle/10665/43048
LinkOut - more resources
Full Text Sources