

Harnessing Brazilian biodiversity database: identification of flavonoids as potential inhibitors of SARS-CoV-2 main protease using computational approaches and all-atom molecular dynamics simulation
- PMID: 38456183
- PMCID: PMC10917896
- DOI: 10.3389/fchem.2024.1336001
Harnessing Brazilian biodiversity database: identification of flavonoids as potential inhibitors of SARS-CoV-2 main protease using computational approaches and all-atom molecular dynamics simulation
Abstract
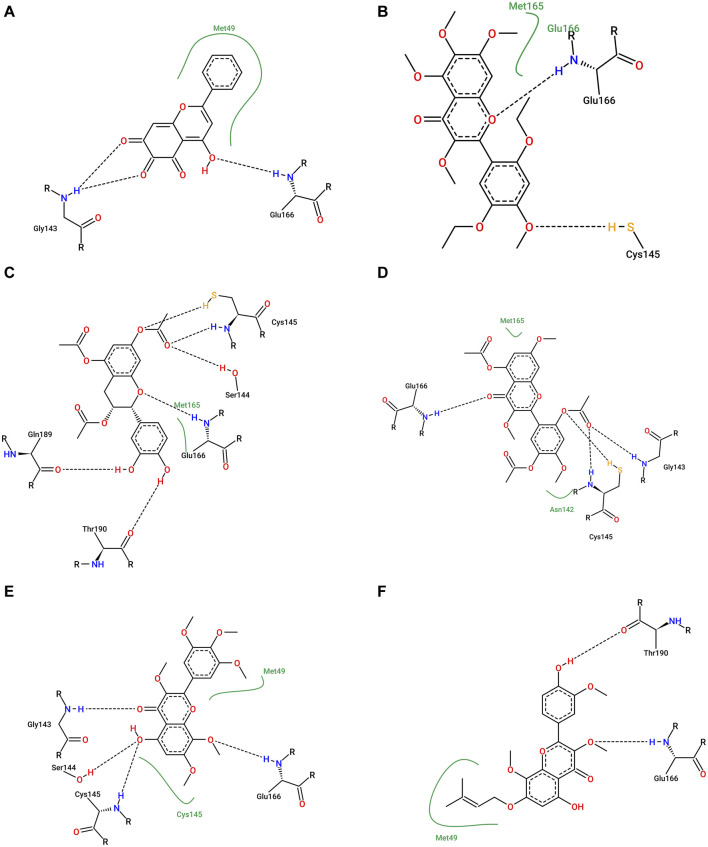
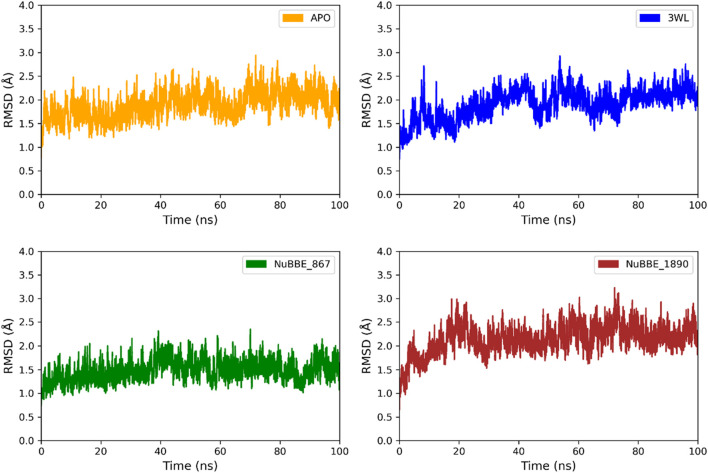
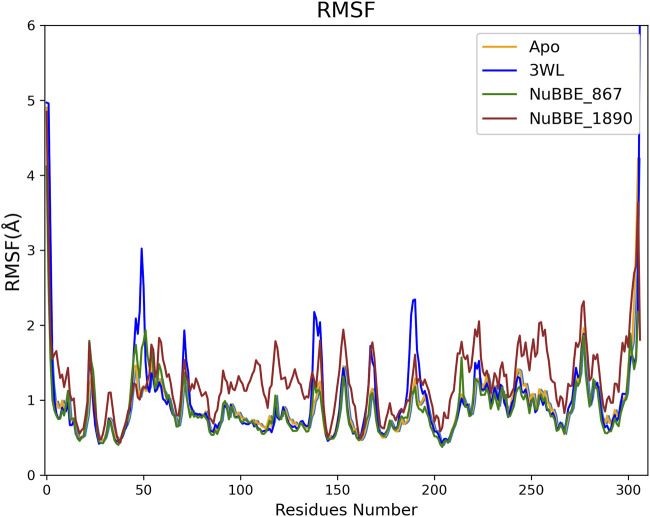
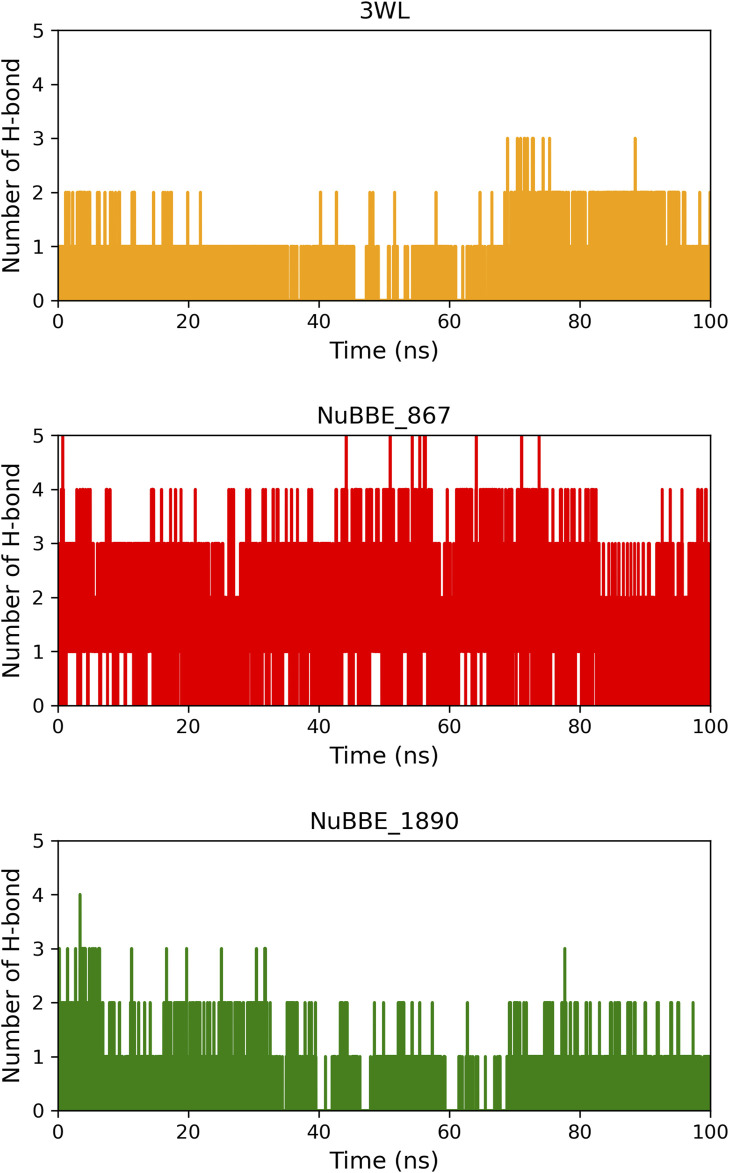
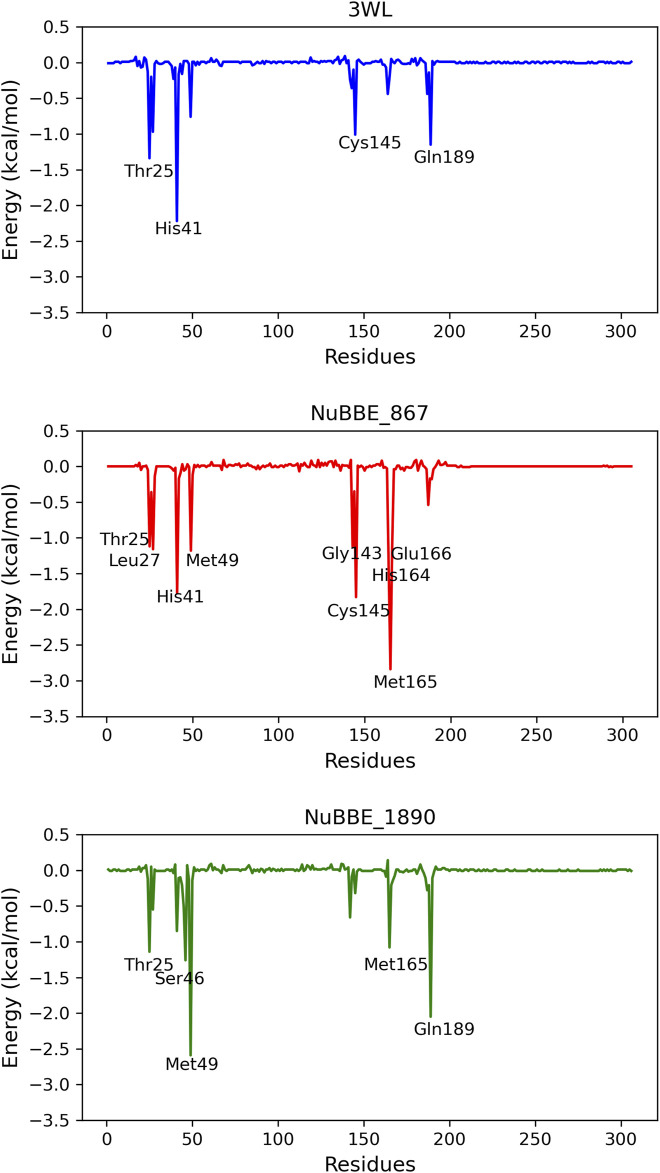
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) is the etiological agent responsible for the global outbreak of COVID-19 (Coronavirus Disease 2019). The main protease of SARS-CoV-2, Mpro, is a key enzyme that plays a vital role in mediating viral replication and transcription. In this study, a comprehensive computational approach was employed to investigate the binding affinity, selectivity, and stability of natural product candidates as potential new antivirals acting on the viral polyprotein processing mediated by SARS-CoV-2 Mpro. A library of 288 flavonoids extracted from Brazilian biodiversity was screened to select potential Mpro inhibitors. An initial filter based on Lipinski's rule of five was applied, and 204 compounds that did not violate any of the Lipinski rules were selected. The compounds were then docked into the active site of Mpro using the GOLD program, and the poses were subsequently re-scored using MM-GBSA (Molecular Mechanics Generalized Born Surface Area) binding free energy calculations performed by AmberTools23. The top five flavonoids with the best MM-GBSA binding free energy values were selected for analysis of their interactions with the active site residues of the protein. Next, we conducted a toxicity and drug-likeness analysis, and non-toxic compounds were subjected to molecular dynamics simulation and free energy calculation using the MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) method. It was observed that the five selected flavonoids had lower MM-GBSA binding free energy with Mpro than the co-crystal ligand. Furthermore, these compounds also formed hydrogen bonds with two important residues, Cys145 and Glu166, in the active site of Mpro. Two compounds that passed the drug-likeness filter showed stable conformations during the molecular dynamics simulations. Among these, NuBBE_867 exhibited the best MM-PBSA binding free energy value compared to the crystallographic inhibitor. Therefore, this study suggests that NuBBE_867 could be a potential inhibitor against the main protease of SARS-CoV-2 and may be further examined to confirm our results.
Keywords: MMPBSA; SARS-CoV-2; drug-likeness; flavonoids; main protease; molecular docking; molecular dynamics; natural products.
Copyright © 2024 da Rocha, da Costa, da Costa, da Rocha, Gomes, Machado, Fagan, Brasil and Lima e Lima.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Some Flavolignans as Potent Sars-Cov-2 Inhibitors via Molecular Docking, Molecular Dynamic Simulations and ADME Analysis.Curr Comput Aided Drug Des. 2022;18(5):337-346. doi: 10.2174/1573409918666220816113516. Curr Comput Aided Drug Des. 2022. PMID: 35975852
-
Computational guided identification of a citrus flavonoid as potential inhibitor of SARS-CoV-2 main protease.Mol Divers. 2021 Aug;25(3):1745-1759. doi: 10.1007/s11030-020-10150-x. Epub 2020 Nov 25. Mol Divers. 2021. PMID: 33236176 Free PMC article.
-
Identification of alkaloids from Justicia adhatoda as potent SARS CoV-2 main protease inhibitors: An in silico perspective.J Mol Struct. 2021 Apr 5;1229:129489. doi: 10.1016/j.molstruc.2020.129489. Epub 2020 Oct 19. J Mol Struct. 2021. PMID: 33100380 Free PMC article.
-
Sterenin M as a potential inhibitor of SARS-CoV-2 main protease identified from MeFSAT database using molecular docking, molecular dynamics simulation and binding free energy calculation.Comput Biol Med. 2021 Aug;135:104568. doi: 10.1016/j.compbiomed.2021.104568. Epub 2021 Jun 12. Comput Biol Med. 2021. PMID: 34174757 Free PMC article.
-
Recent Developments and Applications of the MMPBSA Method.Front Mol Biosci. 2018 Jan 10;4:87. doi: 10.3389/fmolb.2017.00087. eCollection 2017. Front Mol Biosci. 2018. PMID: 29367919 Free PMC article. Review.
Cited by
-
A new paradigm for drug discovery in the treatment of complex diseases: drug discovery and optimization.Chin Med. 2025 Mar 24;20(1):40. doi: 10.1186/s13020-025-01075-4. Chin Med. 2025. PMID: 40122800 Free PMC article. Review.
-
High-Throughput Molecular Modeling and Evaluation of the Anti-Inflammatory Potential of Açaí Constituents against NLRP3 Inflammasome.Int J Mol Sci. 2024 Jul 25;25(15):8112. doi: 10.3390/ijms25158112. Int J Mol Sci. 2024. PMID: 39125681 Free PMC article.
-
Exploring the Anticancer Potential of Lamivudine-Loaded Polymeric Nanoparticles: In Vitro Cytotoxicity, Tissue Deposition, Biochemical Impact In Vivo, and Molecular Simulations Analysis.ACS Appl Bio Mater. 2025 Jun 16;8(6):4815-4828. doi: 10.1021/acsabm.5c00182. Epub 2025 May 19. ACS Appl Bio Mater. 2025. PMID: 40384113 Free PMC article.
-
Resveratrol Alleviates Inflammatory Response Through P2X7/NLRP3 Signaling Pathway: In Silico and In Vitro Evidence from Activated Microglia.Pharmaceuticals (Basel). 2025 Jun 24;18(7):950. doi: 10.3390/ph18070950. Pharmaceuticals (Basel). 2025. PMID: 40732240 Free PMC article.
-
Integration of metabolomics and chemometrics with in-silico and in-vitro approaches to unravel SARS-Cov-2 inhibitors from South African plants.PLoS One. 2025 Mar 26;20(3):e0320415. doi: 10.1371/journal.pone.0320415. eCollection 2025. PLoS One. 2025. PMID: 40138368 Free PMC article.
References
-
- Ahmad S., Abbasi H. W., Shahid S., Gul S., Abbasi S. W. (2021). Molecular docking, simulation and MM-PBSA studies of nigella sativa compounds: a computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn. 39, 4225–4233. 10.1080/07391102.2020.1775129 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous