

Interrogations of single-cell RNA splicing landscapes with SCASL define new cell identities with physiological relevance
- PMID: 38461306
- PMCID: PMC10925056
- DOI: 10.1038/s41467-024-46480-9
Interrogations of single-cell RNA splicing landscapes with SCASL define new cell identities with physiological relevance
Abstract
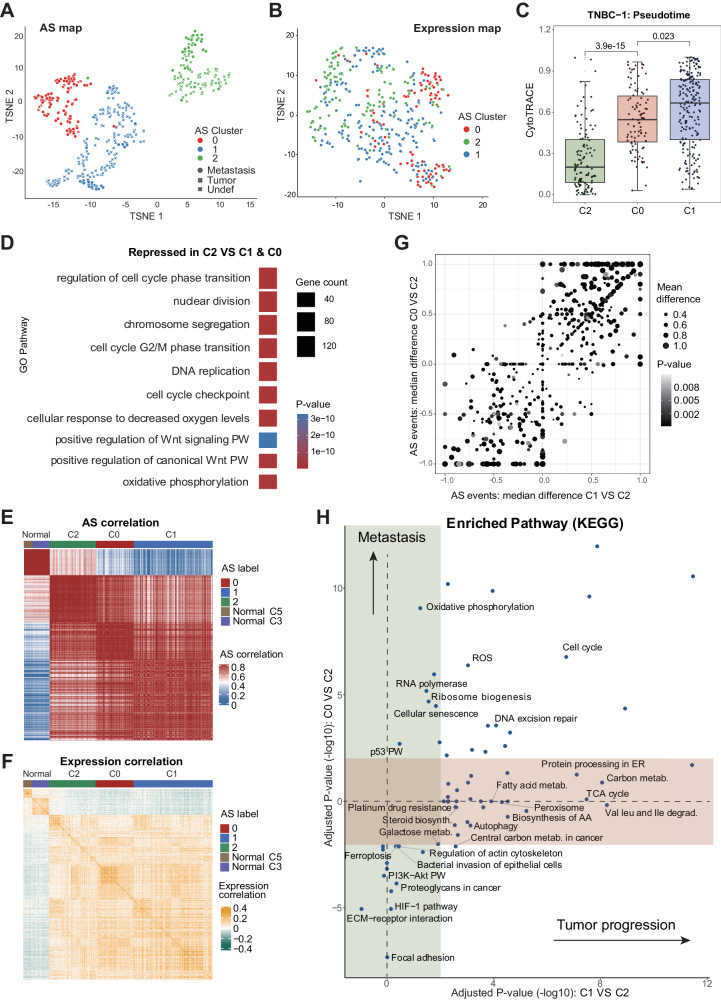
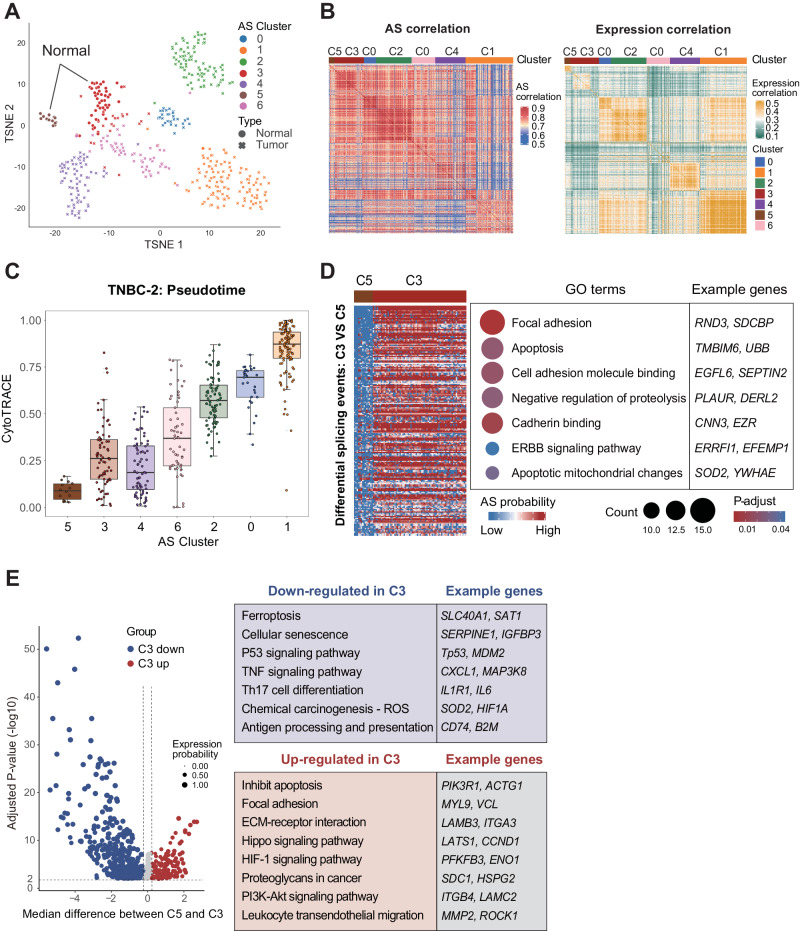
RNA splicing shapes the gene regulatory programs that underlie various physiological and disease processes. Here, we present the SCASL (single-cell clustering based on alternative splicing landscapes) method for interrogating the heterogeneity of RNA splicing with single-cell RNA-seq data. SCASL resolves the issue of biased and sparse data coverage on single-cell RNA splicing and provides a new scheme for classifications of cell identities. With previously published datasets as examples, SCASL identifies new cell clusters indicating potentially precancerous and early-tumor stages in triple-negative breast cancer, illustrates cell lineages of embryonic liver development, and provides fine clusters of highly heterogeneous tumor-associated CD4 and CD8 T cells with functional and physiological relevance. Most of these findings are not readily available via conventional cell clustering based on single-cell gene expression data. Our study shows the potential of SCASL in revealing the intrinsic RNA splicing heterogeneity and generating biological insights into the dynamic and functional cell landscapes in complex tissues.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
