

This is a preprint.
Inhibition of Soluble Epoxide Hydrolase Reduces Inflammation and Myocardial Injury in Arrhythmogenic Cardiomyopathy
- PMID: 38463975
- PMCID: PMC10925075
- DOI: 10.1101/2024.02.17.580812
Inhibition of Soluble Epoxide Hydrolase Reduces Inflammation and Myocardial Injury in Arrhythmogenic Cardiomyopathy
Update in
-
Inhibition of Soluble Epoxide Hydrolase Reduces Inflammation and Myocardial Injury in Arrhythmogenic Cardiomyopathy.JACC Basic Transl Sci. 2025 Mar;10(3):367-380. doi: 10.1016/j.jacbts.2024.12.010. Epub 2025 Feb 5. JACC Basic Transl Sci. 2025. PMID: 40139877 Free PMC article.
Abstract
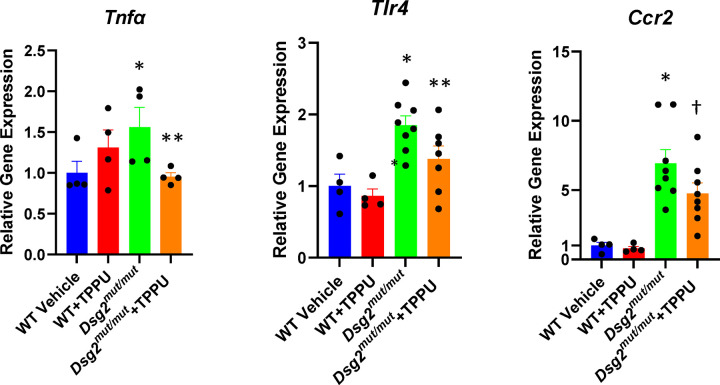
Previous studies have implicated persistent innate immune signaling in the pathogenesis of arrhythmogenic cardiomyopathy (ACM), a familial non-ischemic heart muscle disease characterized by life-threatening arrhythmias and progressive myocardial injury. Here, we provide new evidence implicating inflammatory lipid autocoids in ACM. We show that specialized pro-resolving lipid mediators are reduced in hearts of Dsg2mut/mut mice, a well characterized mouse model of ACM. We also found that ACM disease features can be reversed in rat ventricular myocytes expressing mutant JUP by the pro-resolving epoxy fatty acid (EpFA) 14,15-eicosatrienoic acid (14-15-EET), whereas 14,15-EE-5(Z)E which antagonizes actions of the putative 14,15-EET receptor, intensified nuclear accumulation of the desmosomal protein plakoglobin. Soluble epoxide hydrolase (sEH), an enzyme that rapidly converts pro-resolving EpFAs into polar, far less active or even pro-inflammatory diols, is highly expressed in cardiac myocytes in Dsg2mut/mut mice. Inhibition of sEH prevented progression of myocardial injury in Dsg2mut/mut mice and led to recovery of contractile function. This was associated with reduced myocardial expression of genes involved in the innate immune response and fewer pro-inflammatory macrophages expressing CCR2, which mediate myocardial injury in Dsg2mut/mut mice. These results suggest that pro-inflammatory eicosanoids contribute to the pathogenesis of ACM and, further, that inhibition of sEH may be an effective, mechanism-based therapy for ACM patients.
Conflict of interest statement
Author Conflicts of Interest Disclosures: Dr. Saffitz is a consultant to Implicit Biosciences and Rejuvenate Bio. Dr. Hammock holds patents related to the commercial development of soluble epoxide hydrolase inhibitors for cardiovascular disease. He is Chief Scientific Officer of EicOsis Human Health, currently in human 1b safety trials of the soluble epoxide hydrolase inhibitor EC5026. Dr. Lavine is a consultant for Kiniksa, Cytokinetics, Implicit Biosciences, and SUN Pharmaceuticals. Other authors have no relevant conflicts or financial relationships to disclose.
Figures









References
-
- Corrado D, Link M, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. New Eng J Med, 2017; 376:61–72. - PubMed
-
- Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F. Spectrum of clinico-pathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 1997; 30:1512–1520. - PubMed
-
- Chelko SP, Penna V, Engle M, Landim-Vieira ML, Cannon EN, Lavine K, Saffitz JE. Mechanisms of innate immune injury in arrhythmogenic cardiomyopathy. bioRxiv. 2023. Jul 13.2023.07.12.548682, doi: 10.1101/2023.07.12.548682. - DOI
-
- Hawthorne RN, Blazeski A, Lowenthal J, Kannan S, Teuben R, DiSilvestre D, MorrissetteMcAlmon J, Saffitz JE, Boheler KR, James CA, Chelko SP, Tomaselli G, Tung L. Altered electrical, biomolecular, and immunologic phenotypes in a novel patient-derived stem cell model of desmoglein-2 mutant ARVC. J Clin Med 2021; 10;10(14):3061. doi: 10.3390/jcm10143061. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous