

This is a preprint.
Transcriptome- and proteome-wide effects of a circular RNA encompassing four early exons of the spinal muscular atrophy genes
- PMID: 38464174
- PMCID: PMC10925445
- DOI: 10.21203/rs.3.rs-3818622/v1
Transcriptome- and proteome-wide effects of a circular RNA encompassing four early exons of the spinal muscular atrophy genes
Update in
-
Transcriptome- and proteome-wide effects of a circular RNA encompassing four early exons of the spinal muscular atrophy genes.Sci Rep. 2024 May 7;14(1):10442. doi: 10.1038/s41598-024-60593-7. Sci Rep. 2024. PMID: 38714739 Free PMC article.
Abstract
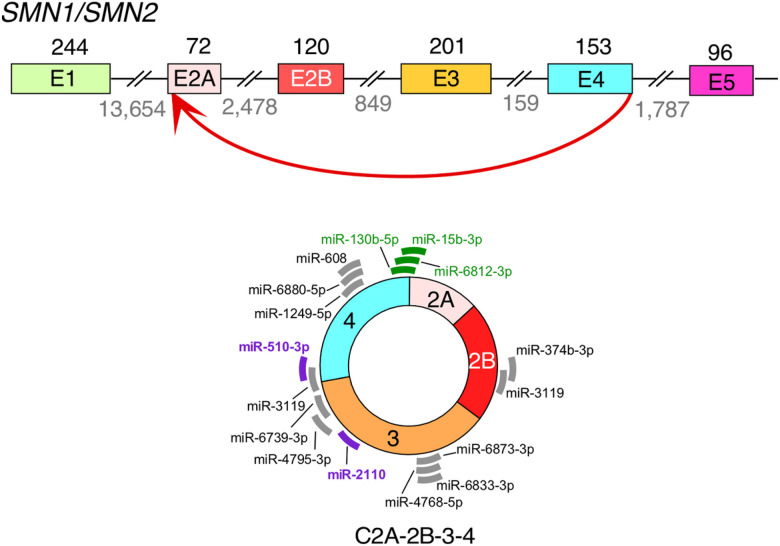
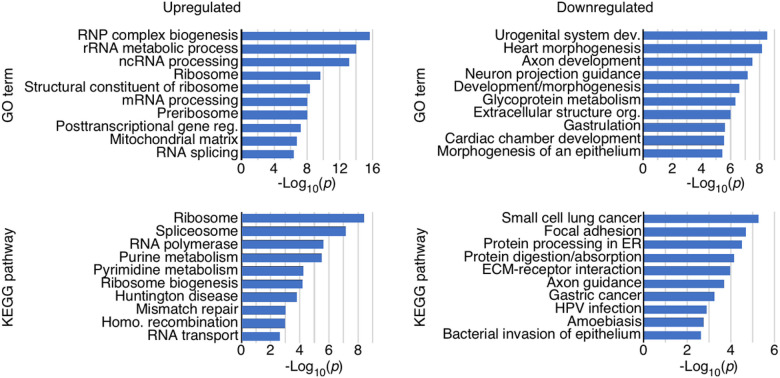
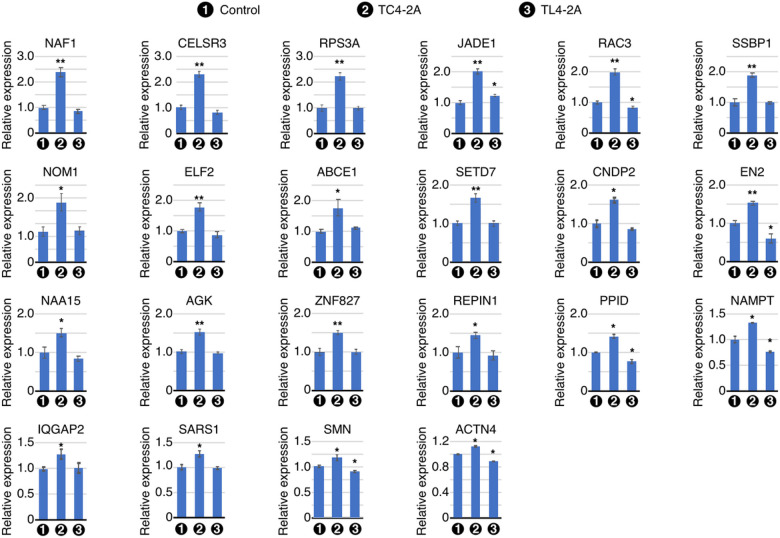
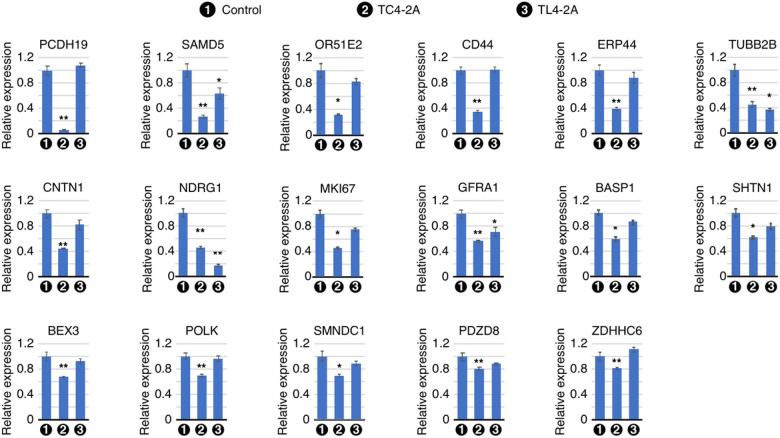
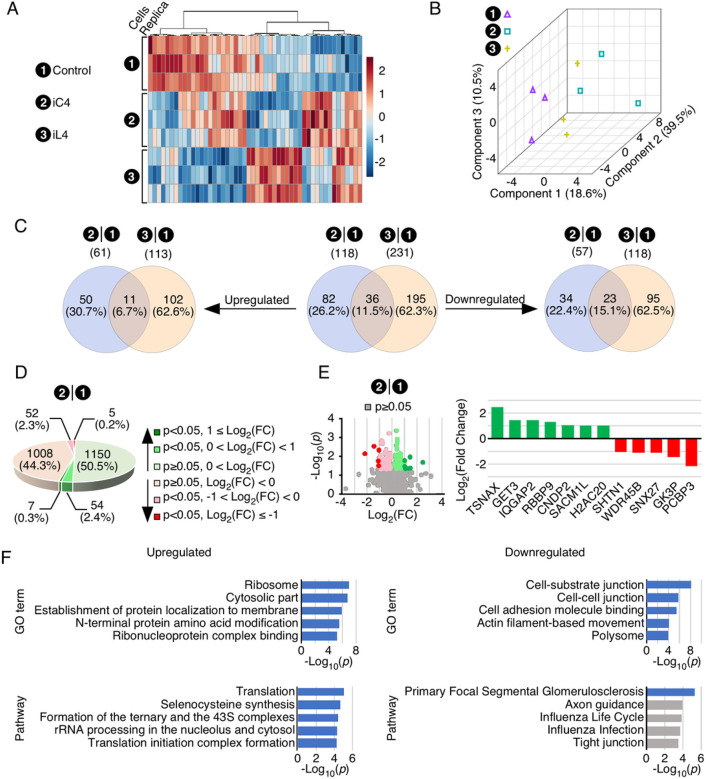
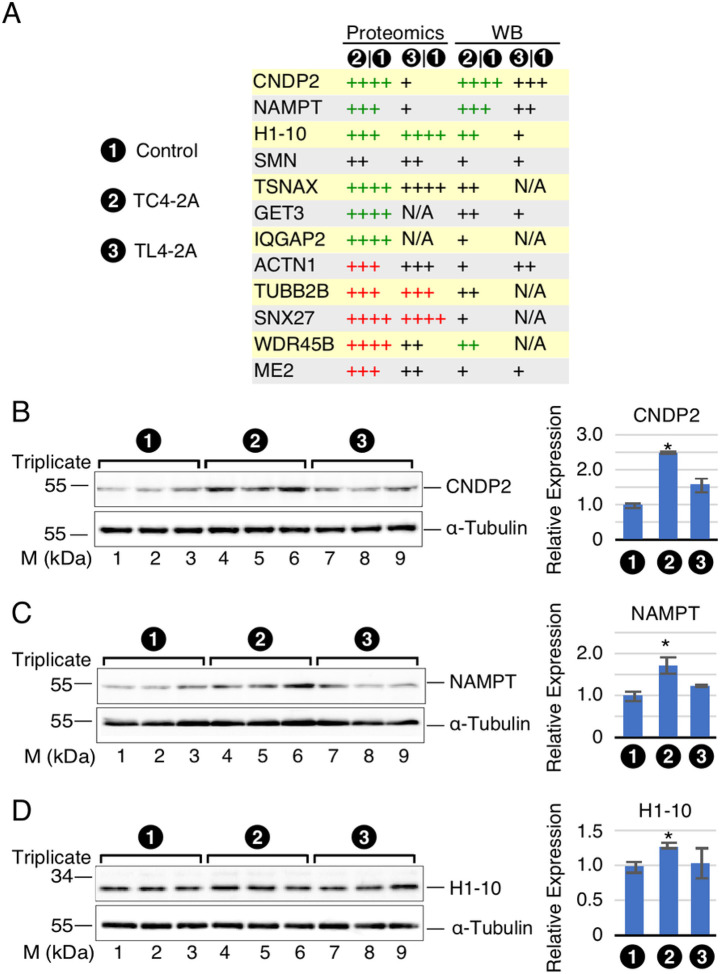
Spinal muscular atrophy (SMA) genes, SMN1 and SMN2, produce multiple circular RNAs (circRNAs), including C2A-2B-3-4 that encompasses early exons 2A, 2B, 3 and 4. Here we report the transcriptome- and proteome-wide effects of overexpression of C2A-2B-3-4 in inducible HEK293 cells. Our RNA-Seq analysis revealed altered expression of ~ 15% genes (4,172 genes) by C2A-2B-3-4. About half of the affected genes by C2A-2B-3-4 remained unaffected by L2A-2B-3-4, a linear transcript encompassing exons 2A, 2B, 3 and 4 of SMN1/SMN2. These fifindings underscore the unique role of the structural context of C2A-2B-3-4 in gene regulation. A surprisingly high number of upregulated genes by C2A-2B-3-4 were located on chromosomes 4 and 7, whereas many of the downregulated genes were located on chromosomes 10 and X. Supporting a cross-regulation of SMN1/SMN2 transcripts, C2A-2B-3-4 and L2A-2B-3-4 upregulated and downregulated SMN1/SMN2 mRNAs, respectively. Proteome analysis revealed 61 upregulated and 57 downregulated proteins by C2A-2B-3-4 with very limited overlap with those affected by L2A-2B-3-4. Independent validations confirmed the effect of C2A-2B-3-4 on expression of genes associated with chromatin remodeling, transcription, spliceosome function, ribosome biogenesis, lipid metabolism, cytoskeletal formation, cell proliferation and neuromuscular junction formation. Our findings reveal a broad role of C2A-2B-3-4, a universally expressed circRNA produced by SMN1/SMN2.
Keywords: SMA; SMN; Survival Motor Neuron; circRNA; circular RNA; proteome; spinal muscular atrophy; transcriptome.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures

Similar articles
-
Transcriptome- and proteome-wide effects of a circular RNA encompassing four early exons of the spinal muscular atrophy genes.Sci Rep. 2024 May 7;14(1):10442. doi: 10.1038/s41598-024-60593-7. Sci Rep. 2024. PMID: 38714739 Free PMC article.
-
Internal Introns Promote Backsplicing to Generate Circular RNAs from Spinal Muscular Atrophy Gene.Genes (Basel). 2022 Jun 25;13(7):1145. doi: 10.3390/genes13071145. Genes (Basel). 2022. PMID: 35885927 Free PMC article.
-
Revealing diverse alternative splicing variants of the highly homologous SMN1 and SMN2 genes by targeted long-read sequencing.Mol Genet Genomics. 2022 Jul;297(4):1039-1048. doi: 10.1007/s00438-022-01874-6. Epub 2022 May 25. Mol Genet Genomics. 2022. PMID: 35612622
-
Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development.Int J Mol Sci. 2021 Jul 23;22(15):7896. doi: 10.3390/ijms22157896. Int J Mol Sci. 2021. PMID: 34360669 Free PMC article. Review.
-
Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases.Front Mol Biosci. 2016 Mar 10;3:7. doi: 10.3389/fmolb.2016.00007. eCollection 2016. Front Mol Biosci. 2016. PMID: 27014701 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
