

This is a preprint.
Domain coupling in activation of a family C GPCR
- PMID: 38464305
- PMCID: PMC10925283
- DOI: 10.1101/2024.02.28.582567
Domain coupling in activation of a family C GPCR
Update in
-
Domain coupling in activation of a family C GPCR.Nat Chem Biol. 2025 Sep;21(9):1433-1443. doi: 10.1038/s41589-025-01895-3. Epub 2025 Apr 25. Nat Chem Biol. 2025. PMID: 40281344 Free PMC article.
Abstract
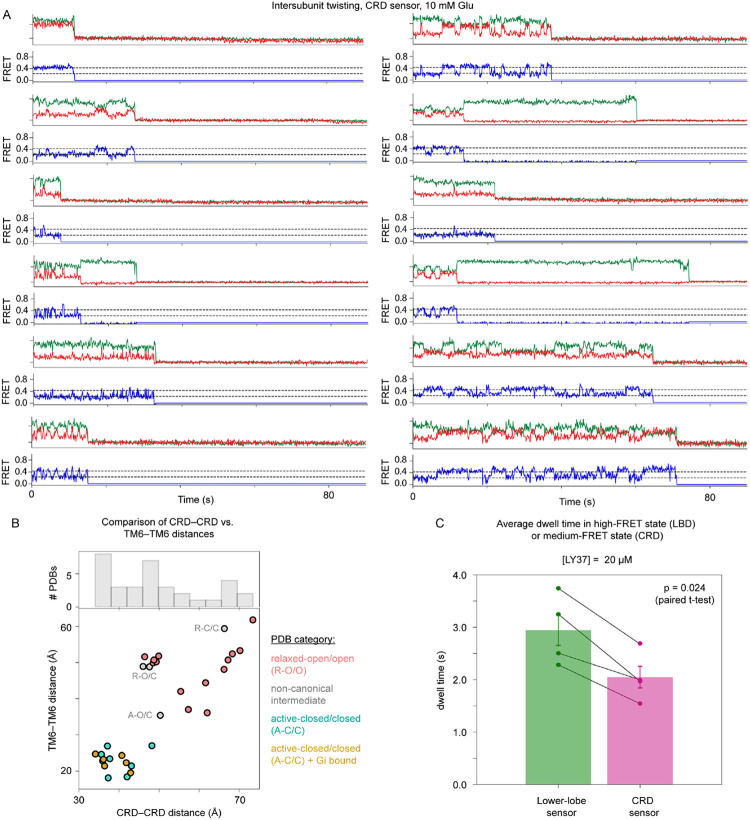
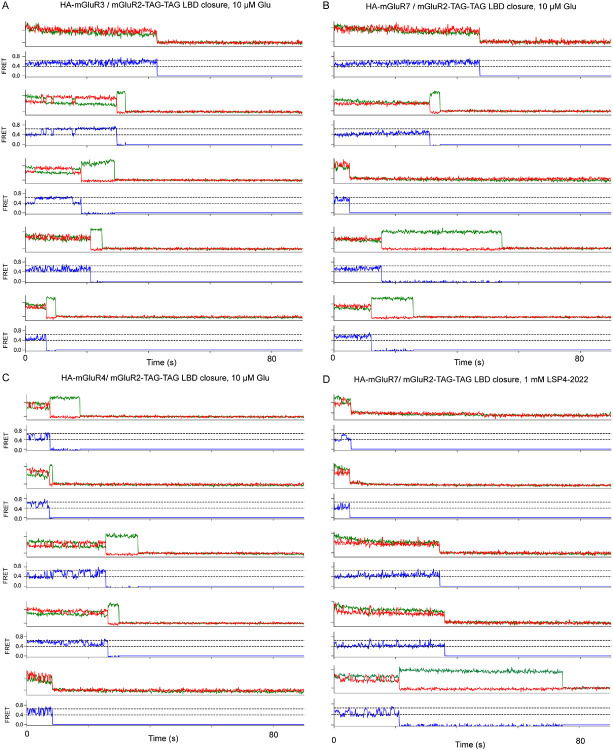
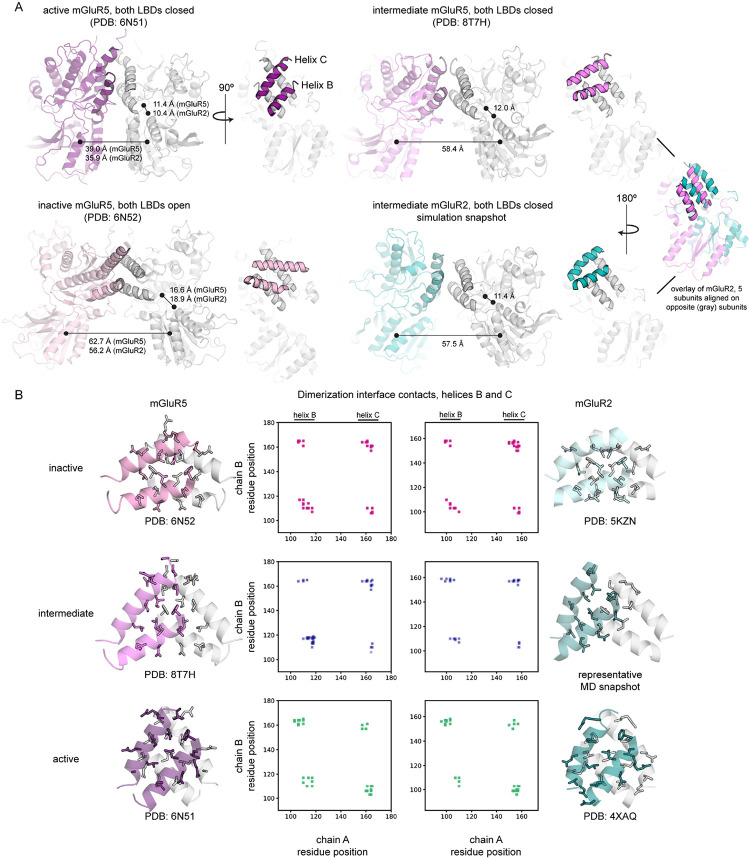
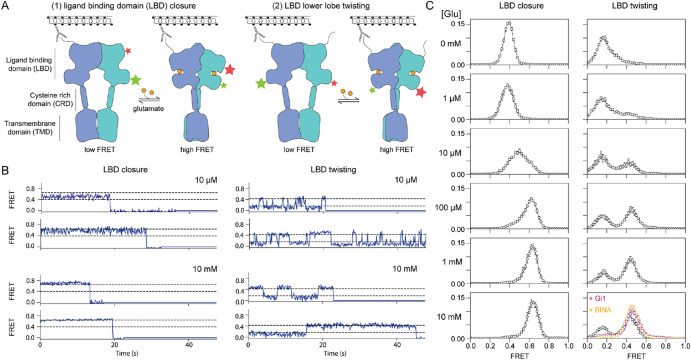
The G protein-coupled metabotropic glutamate receptors form homodimers and heterodimers with highly diverse responses to glutamate and varying physiological function. The molecular basis for this diversity remains poorly delineated. We employ molecular dynamics, single-molecule spectroscopy, and hydrogen-deuterium exchange to dissect the pathway of activation triggered by glutamate. We find that activation entails multiple loosely coupled steps and identify a novel pre-active intermediate whose transition to the active state forms dimer interactions that set signaling efficacy. Such subunit interactions generate functional diversity that differs across homodimers and heterodimers. The agonist-bound receptor is remarkably dynamic, with low occupancy of G protein-coupling conformations, providing considerable headroom for modulation of the landscape by allosteric ligands. Sites of sequence diversity within the dimerization interface and diverse coupling between activation rearrangements may contribute to precise decoding of glutamate signals and transients over broad spatial and temporal scales.
Figures








References
-
- T. S. F. Hansen Kasper B., Wollmuth Lonnie P., Bowie Derek, Furukawa Hiro, Menniti Frank S., Sobolevsky Alexander I., Swanson Geoffrey T., Swanger Sharon A., Greger Ingo H., Nakagawa Terunaga, McBain Chris J., Jayaraman Vasanthi, Low Chian-Ming, Mark L. Dell’Acqu, Structure, Function and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 73, 298–487 (2021). - PMC - PubMed
-
- Koehl A., Hu H., Feng D., Sun B., Zhang Y., Robertson M. J., Chu M., Kobilka T. S., Laermans T., Steyaert J., Tarrasch J., Dutta S., Fonseca R., Weis W. I., Mathiesen J. M., Skiniotis G., Kobilka B. K., Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019). - PMC - PubMed
-
- Du J., Wang D., Fan H., Xu C., Tai L., Lin S., Han S., Tan Q., Wang X., Xu T., Zhang H., Chu X., Yi C., Liu P., Wang X., Zhou Y., Pin J. P., Rondard P., Liu H., Liu J., Sun F., Wu B., Zhao Q., Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 594 (2021). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials