

This is a preprint.
Ribonanza: deep learning of RNA structure through dual crowdsourcing
- PMID: 38464325
- PMCID: PMC10925082
- DOI: 10.1101/2024.02.24.581671
Ribonanza: deep learning of RNA structure through dual crowdsourcing
Abstract
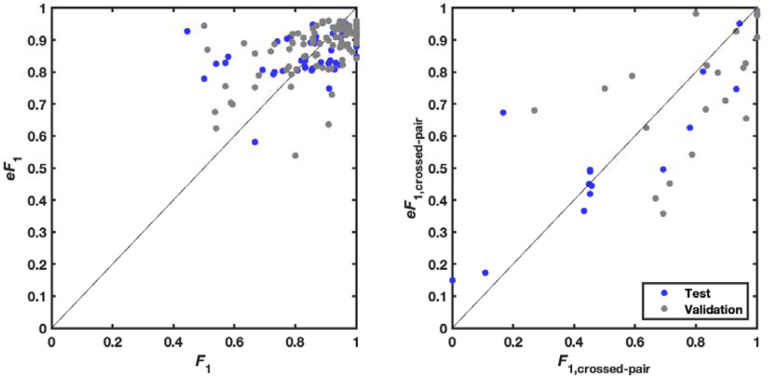
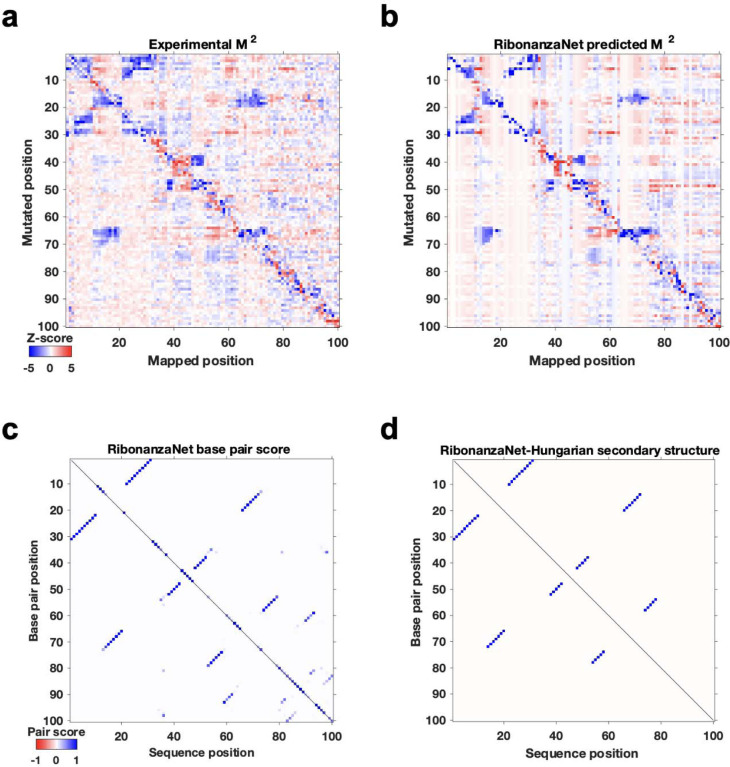
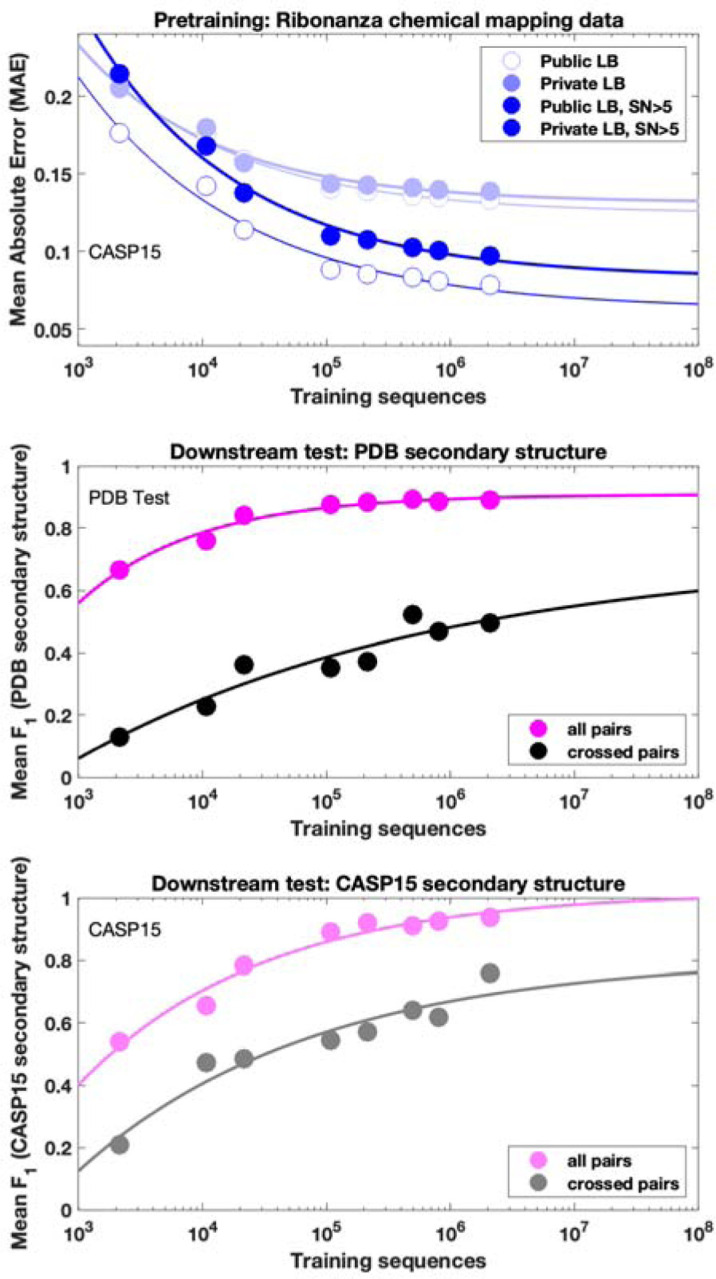
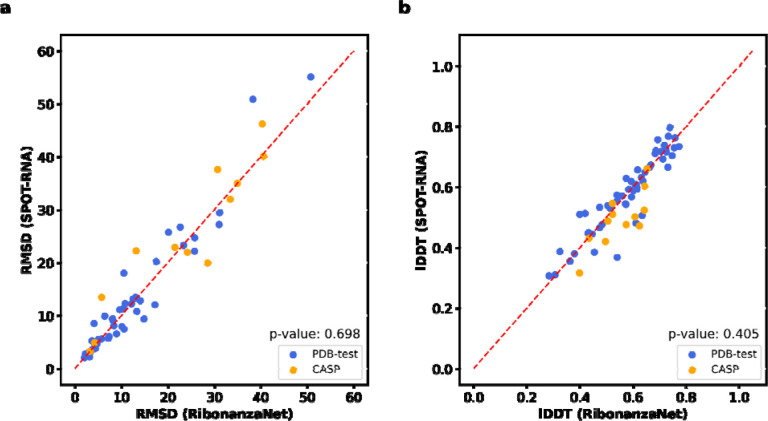
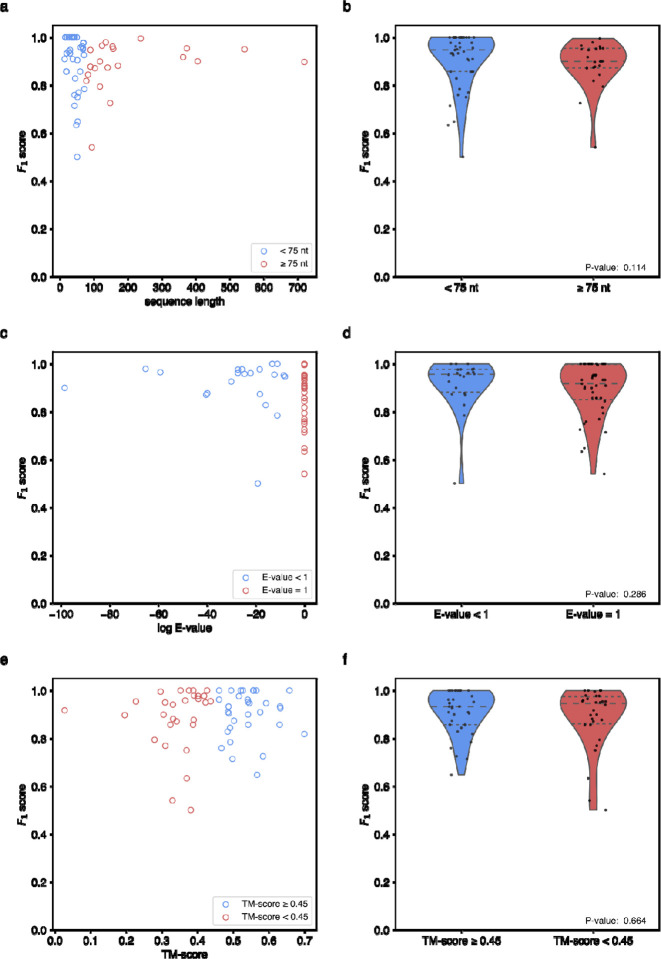
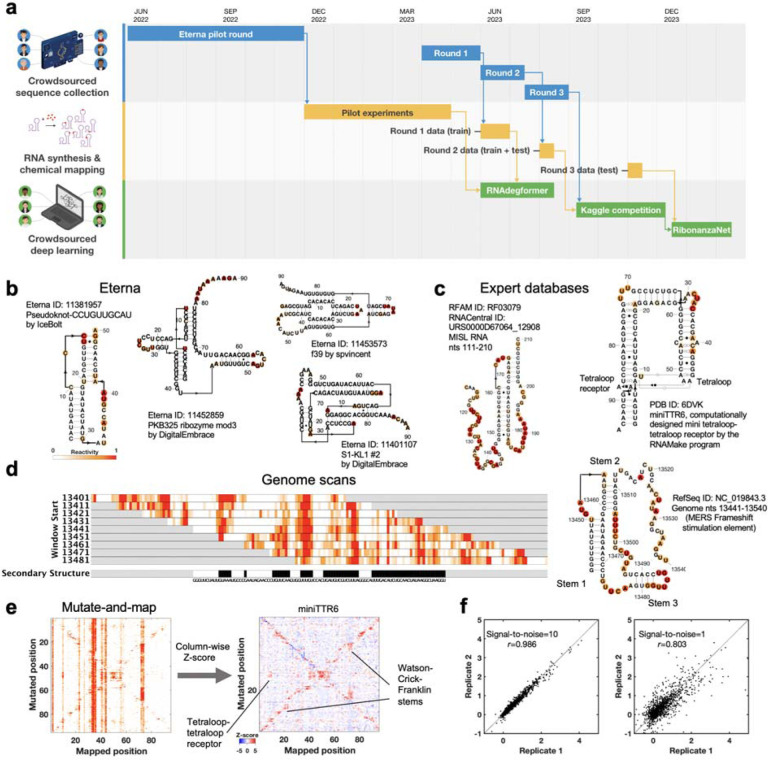
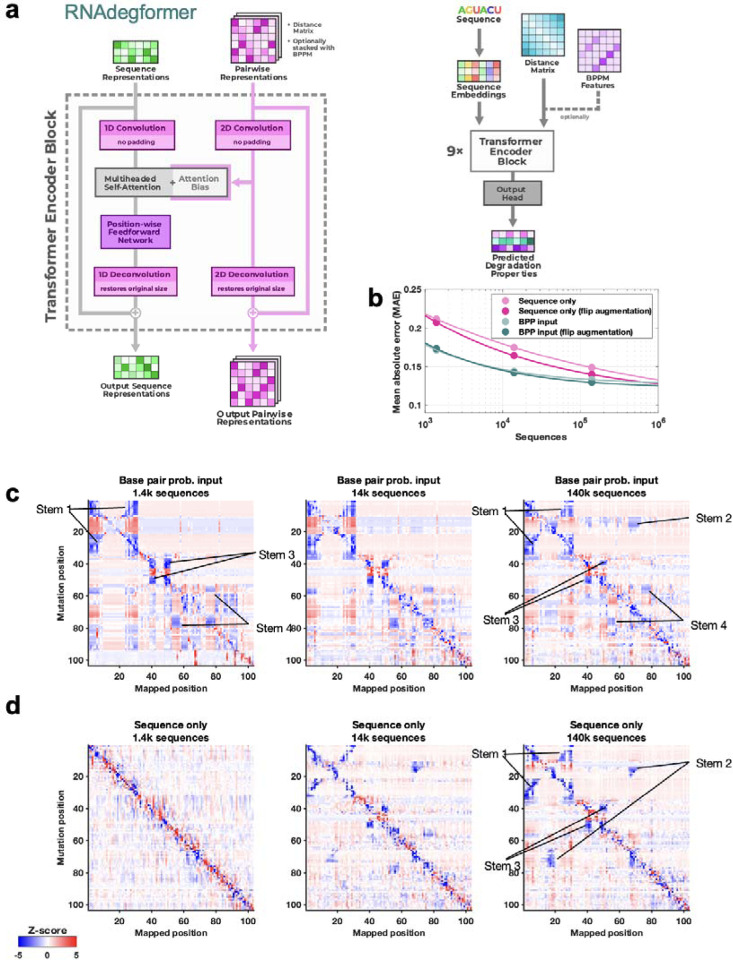
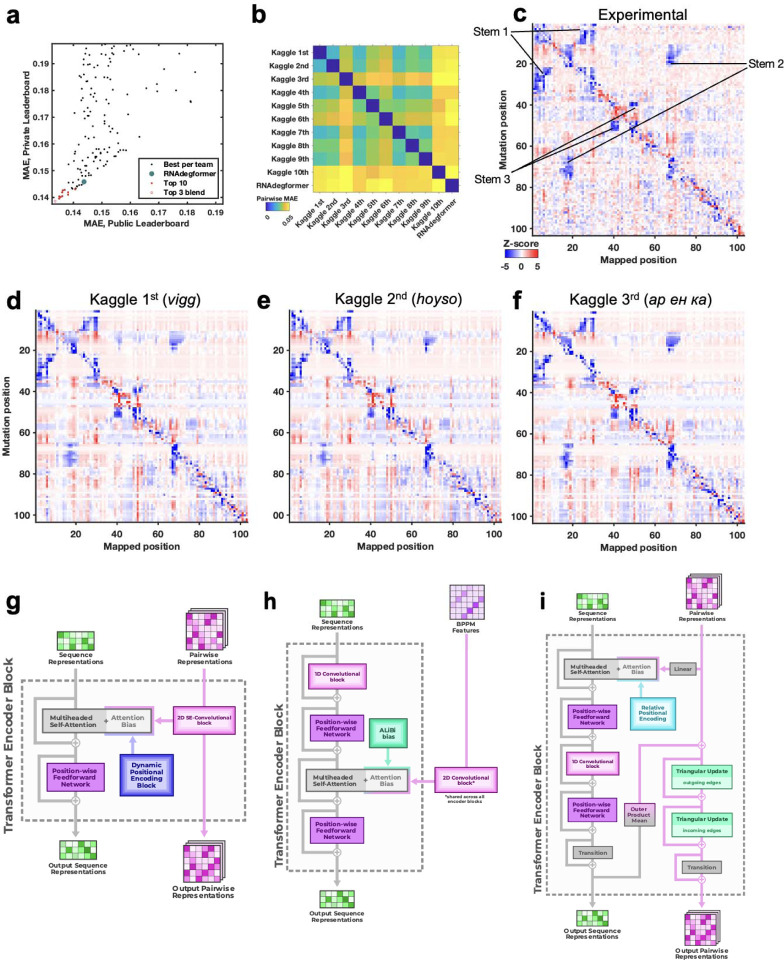
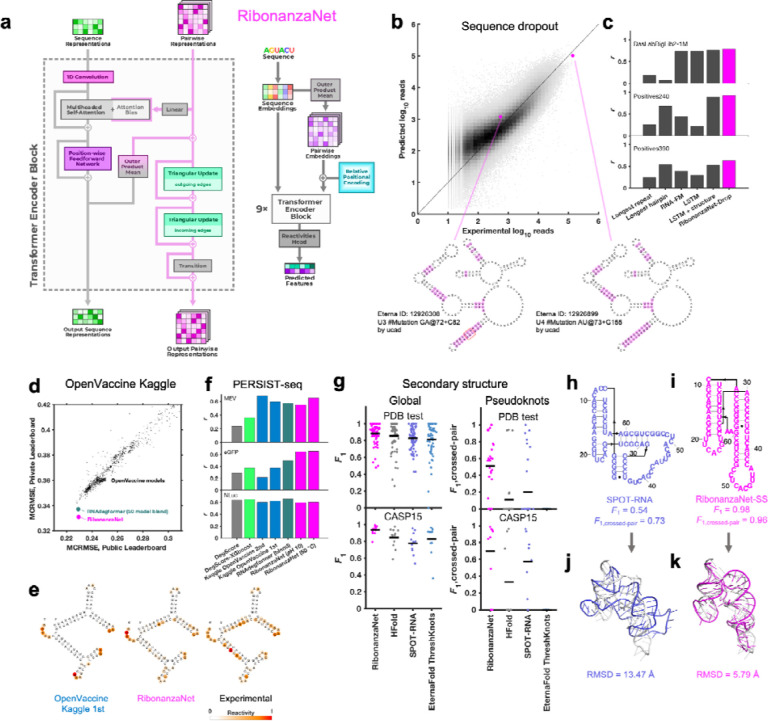
Prediction of RNA structure from sequence remains an unsolved problem, and progress has been slowed by a paucity of experimental data. Here, we present Ribonanza, a dataset of chemical mapping measurements on two million diverse RNA sequences collected through Eterna and other crowdsourced initiatives. Ribonanza measurements enabled solicitation, training, and prospective evaluation of diverse deep neural networks through a Kaggle challenge, followed by distillation into a single, self-contained model called RibonanzaNet. When fine tuned on auxiliary datasets, RibonanzaNet achieves state-of-the-art performance in modeling experimental sequence dropout, RNA hydrolytic degradation, and RNA secondary structure, with implications for modeling RNA tertiary structure.
Figures
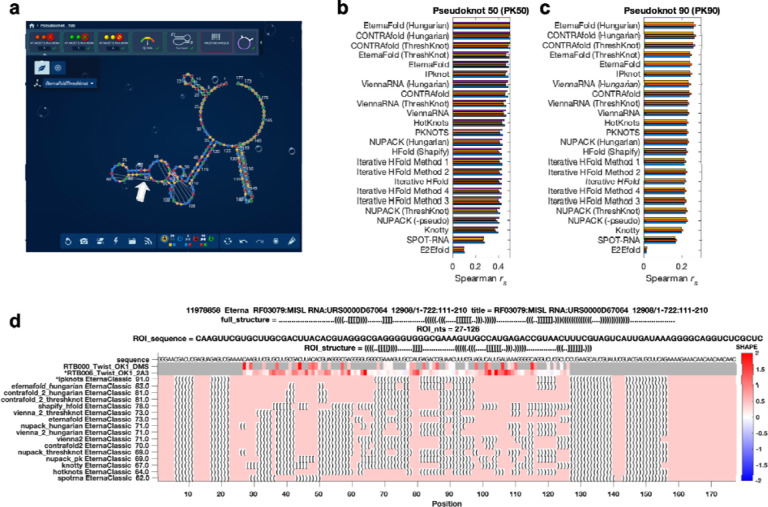
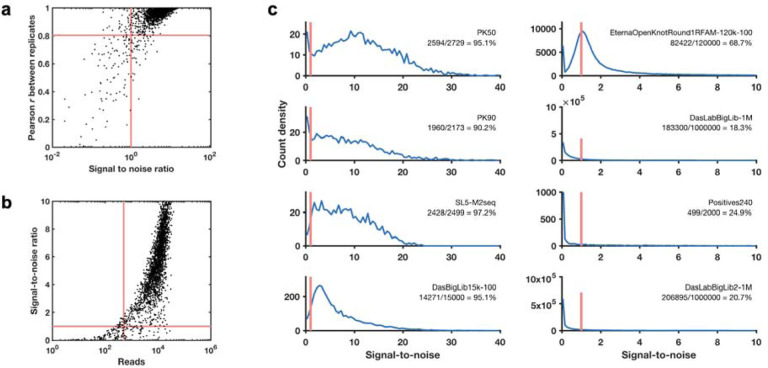
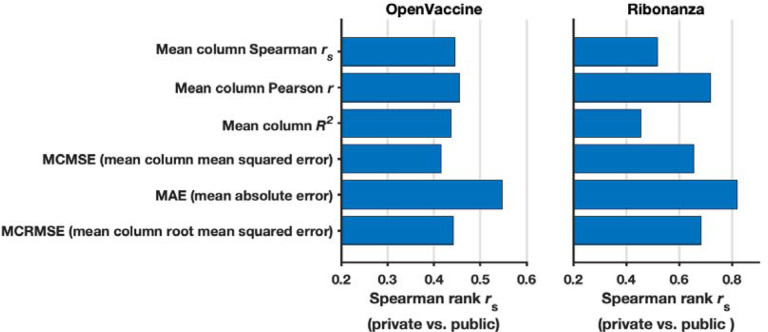
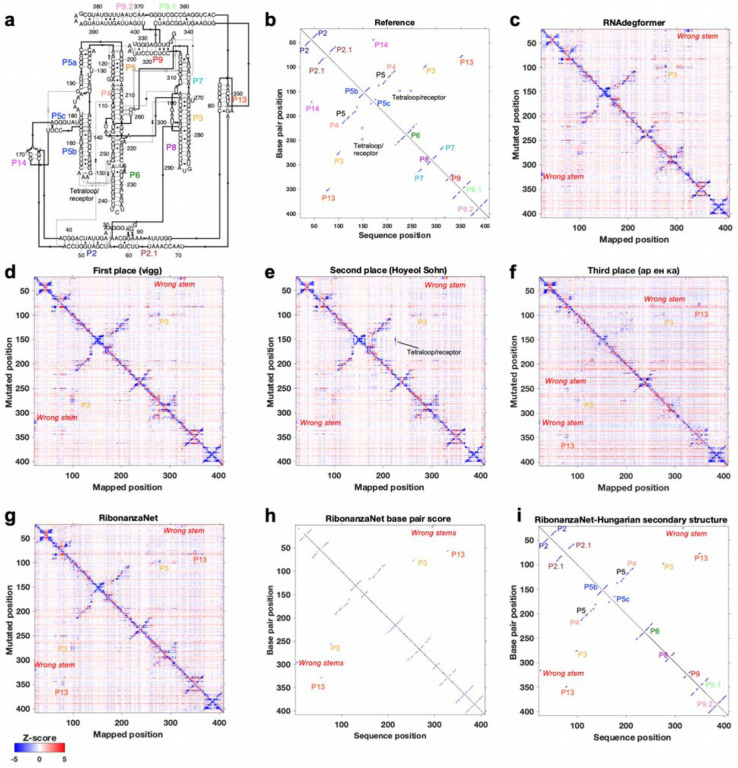
References
-
- Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., Bridgland A., Meyer C., Kohl S. A. A., Ballard A. J., Cowie A., Romera-Paredes B., Nikolov S., Jain R., Adler J., Back T., Petersen S., Reiman D., Clancy E., Zielinski M., Steinegger M., Pacholska M., Berghammer T., Bodenstein S., Silver D., Vinyals O., Senior A. W., Kavukcuoglu K., Kohli P. & Hassabis D. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). - PMC - PubMed
-
- Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G. R., Wang J., Cong Q., Kinch L. N., Schaeffer R. D., Millán C., Park H., Adams C., Glassman C. R., DeGiovanni A., Pereira J. H., Rodrigues A. V., van Dijk A. A., Ebrecht A. C., Opperman D. J., Sagmeister T., Buhlheller C., Pavkov-Keller T., Rathinaswamy M. K., Dalwadi U., Yip C. K., Burke J. E., Garcia K. C., Grishin N. V., Adams P. D., Read R. J. & Baker D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021). - PMC - PubMed
-
- Chen K., Zhou Y., Wang S. & Xiong P. RNA tertiary structure modeling with BRiQ potential in CASP15. Proteins 91, 1771–1778 (2023). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources