

Endothelial Slc35a1 Deficiency Causes Loss of LSEC Identity and Exacerbates Neonatal Lipid Deposition in the Liver in Mice
- PMID: 38467191
- PMCID: PMC11061248
- DOI: 10.1016/j.jcmgh.2024.03.002
Endothelial Slc35a1 Deficiency Causes Loss of LSEC Identity and Exacerbates Neonatal Lipid Deposition in the Liver in Mice
Abstract
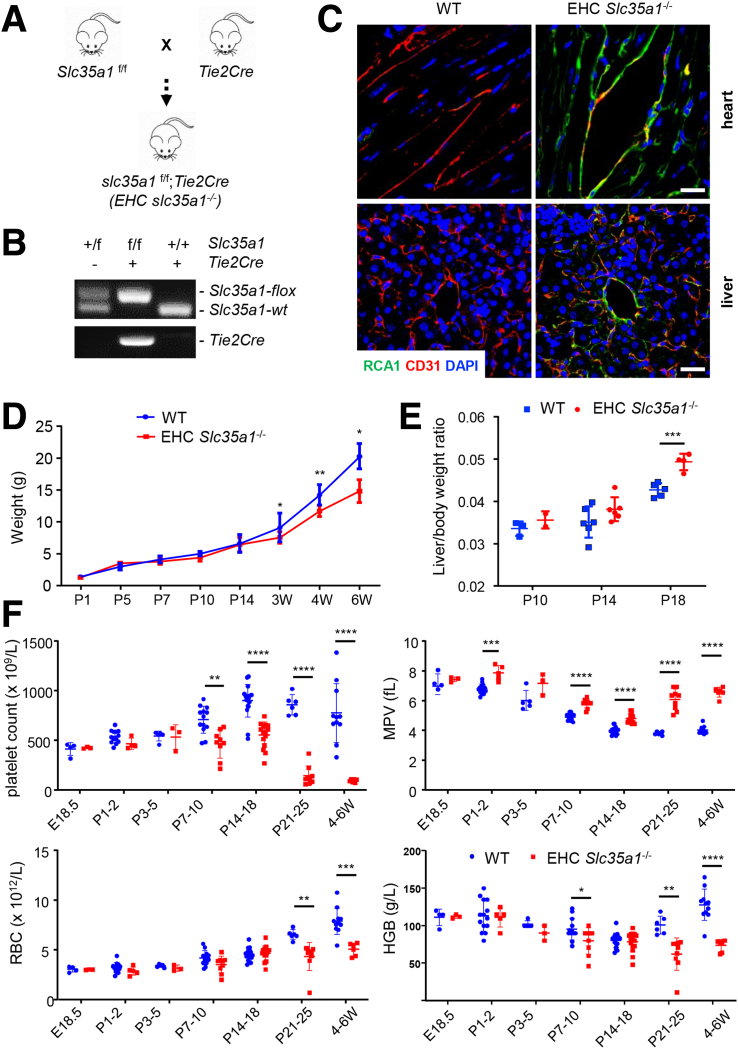
Background & aims: The functional maturation of the liver largely occurs after birth. In the early stages of life, the liver of a newborn encounters enormous high-fat metabolic stress caused by the consumption of breast milk. It is unclear how the maturing liver adapts to high lipid metabolism. Liver sinusoidal endothelial cells (LSECs) play a fundamental role in establishing liver vasculature and are decorated with many glycoproteins on their surface. The Slc35a1 gene encodes a cytidine-5'-monophosphate (CMP)-sialic acid transporter responsible for transporting CMP-sialic acids between the cytoplasm and the Golgi apparatus for protein sialylation. This study aimed to determine whether endothelial sialylation plays a role in hepatic vasculogenesis and functional maturation.
Methods: Endothelial-specific Slc35a1 knockout mice were generated. Liver tissues were collected for histologic analysis, lipidomic profiling, RNA sequencing, confocal immunofluorescence, and immunoblot analyses.
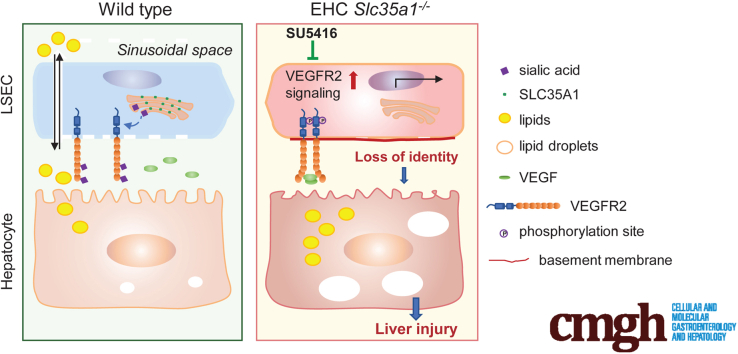
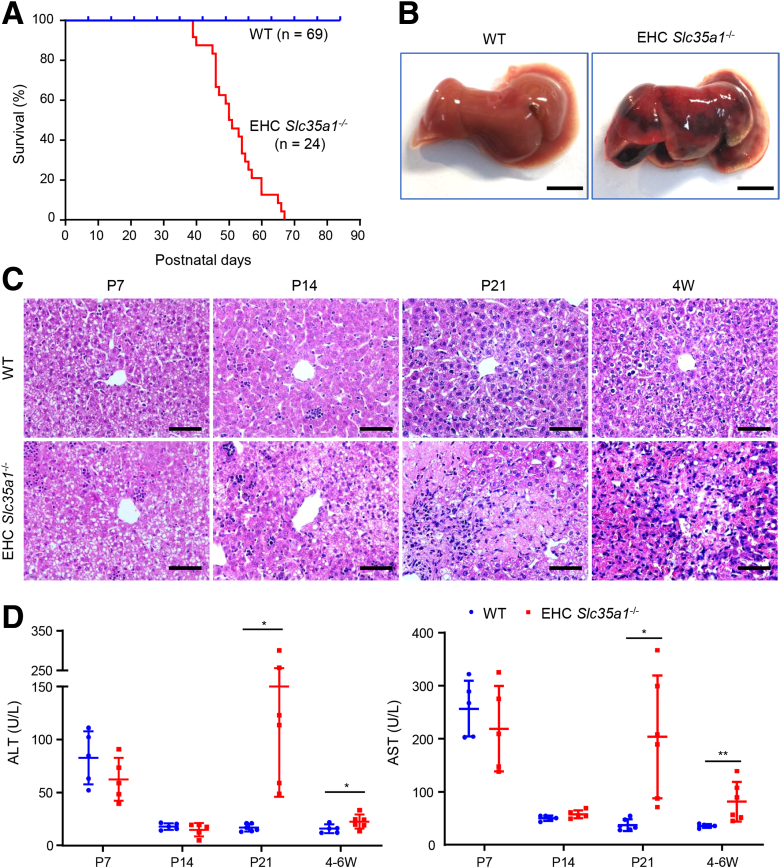
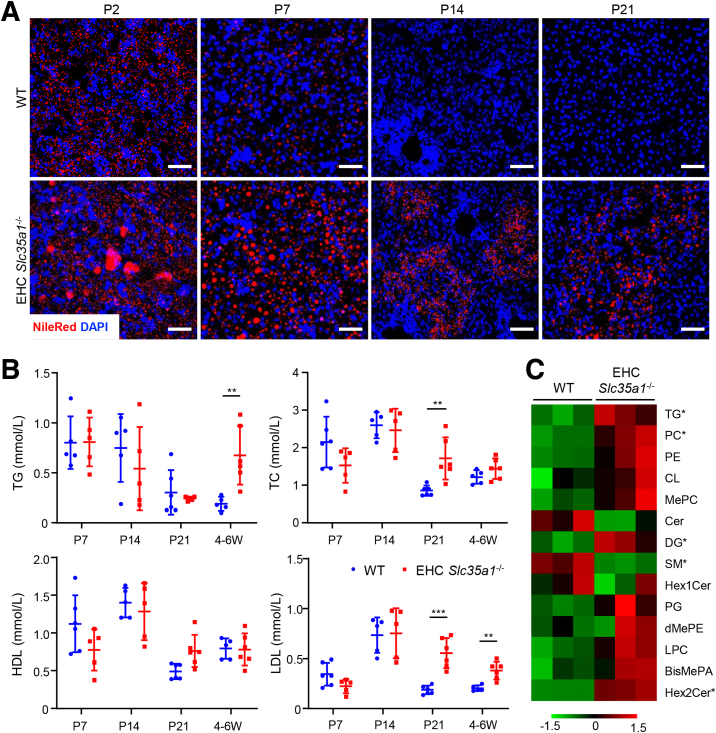
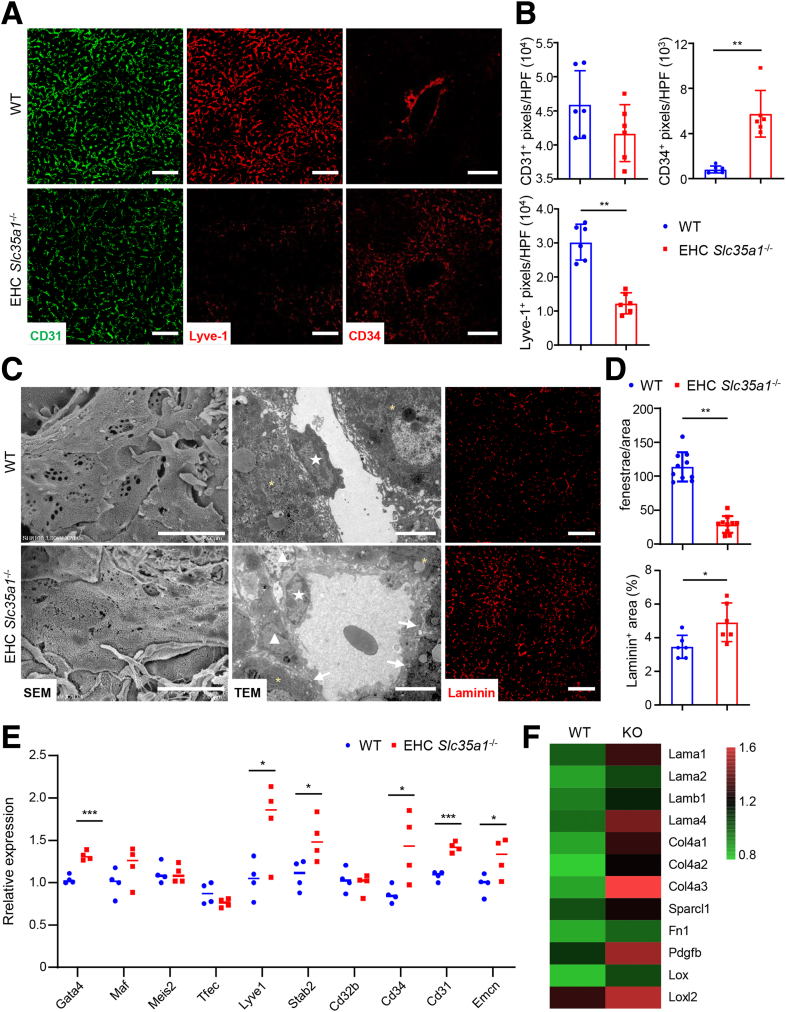
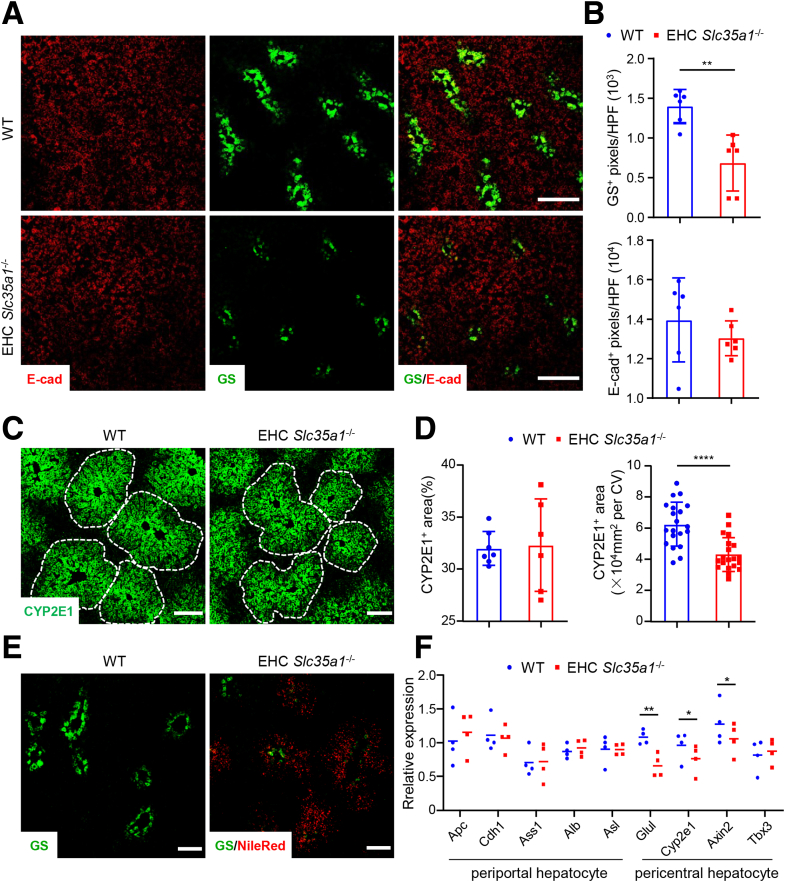
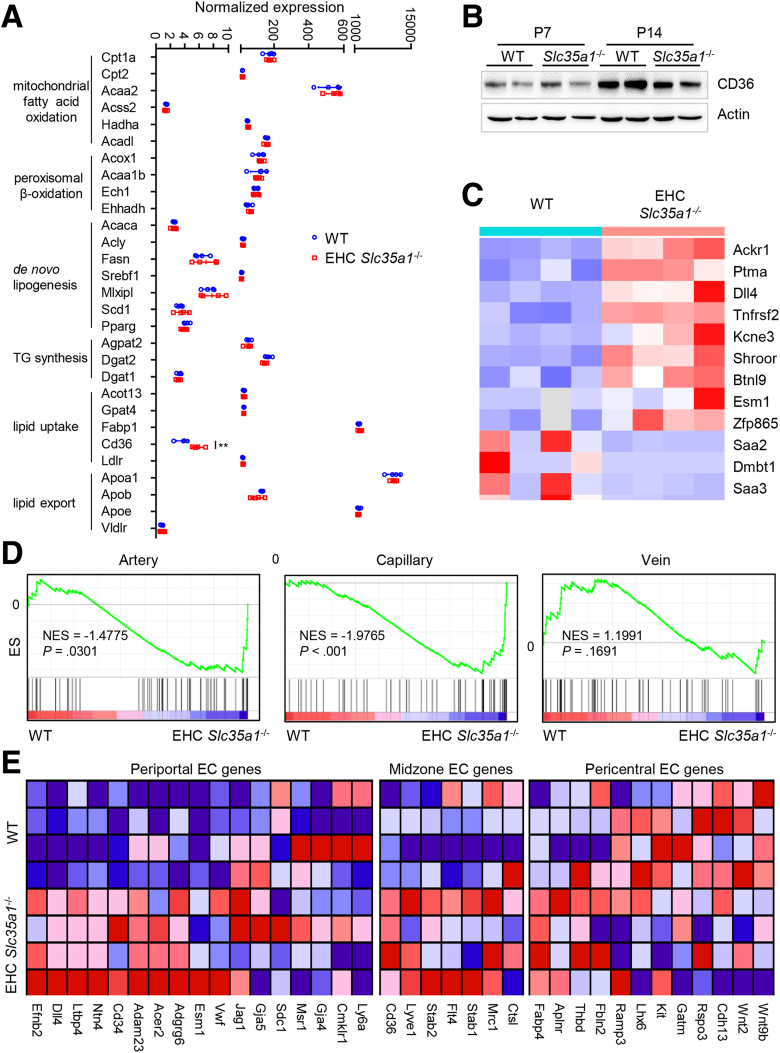
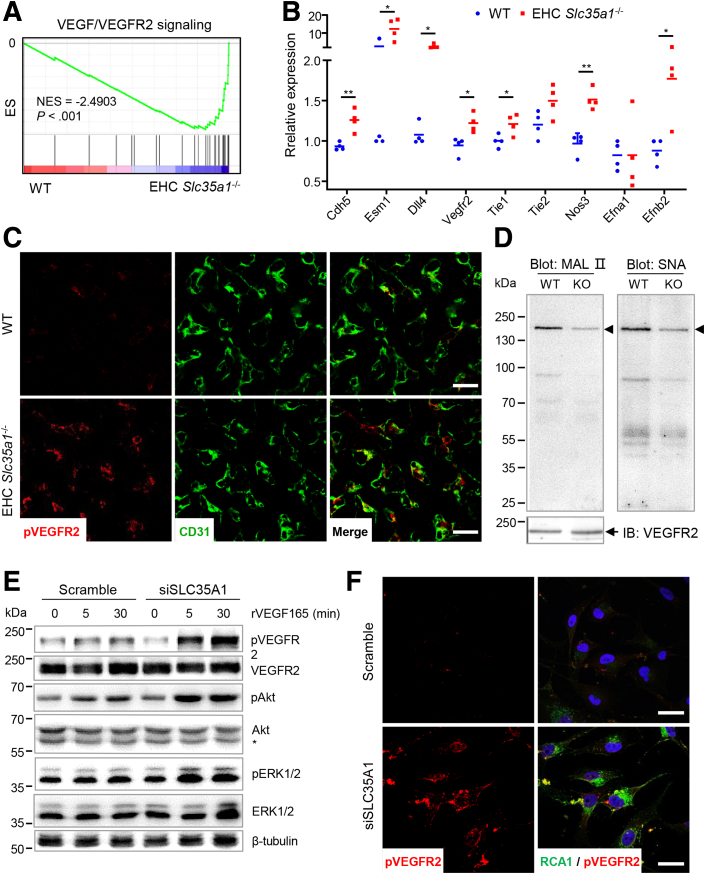
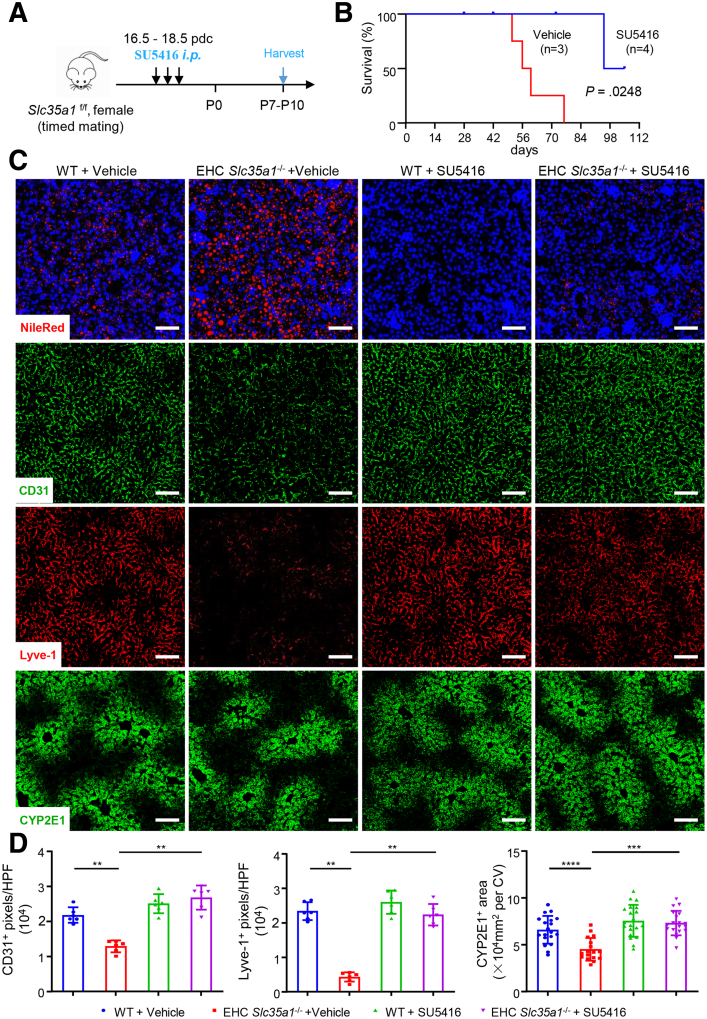
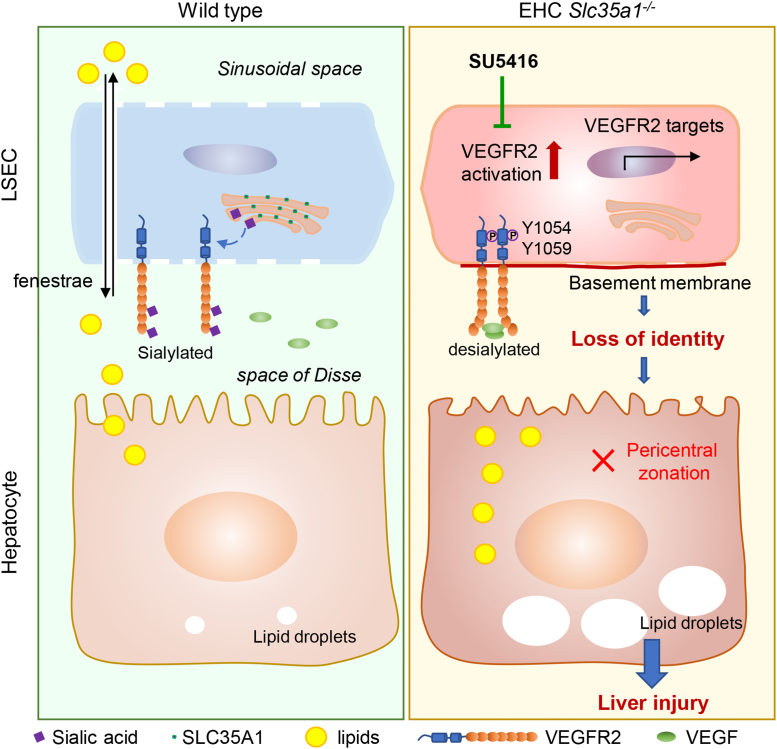
Results: Endothelial Slc35a1-deficient mice exhibited excessive neonatal hepatic lipid deposition, severe liver damage, and high mortality. Endothelial deletion of Slc35a1 led to sinusoidal capillarization and disrupted hepatic zonation. Mechanistically, vascular endothelial growth factor receptor 2 (VEGFR2) in LSECs was desialylated and VEGFR2 signaling was enhanced in Slc35a1-deficient mice. Inhibition of VEGFR2 signaling by SU5416 alleviated lipid deposition and restored hepatic vasculature in Slc35a1-deficient mice.
Conclusions: Our findings suggest that sialylation of LSECs is critical for maintaining hepatic vascular development and lipid homeostasis. Targeting VEGFR2 signaling may be a new strategy to prevent liver disorders associated with abnormal vasculature and lipid deposition.
Keywords: Lipid Deposition; Liver Injury; Liver Sinusoidal Endothelial Cell; Slc35a1; VEGFR2.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Slc35a1 deficiency causes thrombocytopenia due to impaired megakaryocytopoiesis and excessive platelet clearance in the liver.Haematologica. 2021 Mar 1;106(3):759-769. doi: 10.3324/haematol.2019.225987. Haematologica. 2021. PMID: 32303557 Free PMC article.
-
Identification of SLC35A1 as an essential host factor for the transduction of multi-serotype recombinant adeno-associated virus (AAV) vectors.mBio. 2025 Jan 8;16(1):e0326824. doi: 10.1128/mbio.03268-24. Epub 2024 Nov 27. mBio. 2025. PMID: 39601564 Free PMC article.
-
Disease mutations in CMP-sialic acid transporter SLC35A1 result in abnormal α-dystroglycan O-mannosylation, independent from sialic acid.Hum Mol Genet. 2015 Apr 15;24(8):2241-6. doi: 10.1093/hmg/ddu742. Epub 2014 Dec 30. Hum Mol Genet. 2015. PMID: 25552652
-
Liver sinusoidal endothelial cells are implicated in multiple fibrotic mechanisms.Mol Biol Rep. 2021 Mar;48(3):2803-2815. doi: 10.1007/s11033-021-06269-1. Epub 2021 Mar 17. Mol Biol Rep. 2021. PMID: 33730288 Review.
-
Liver Sinusoidal Endothelial Cells at the Crossroad of Iron Overload and Liver Fibrosis.Antioxid Redox Signal. 2021 Aug 20;35(6):474-486. doi: 10.1089/ars.2020.8168. Epub 2020 Aug 27. Antioxid Redox Signal. 2021. PMID: 32689808 Review.
Cited by
-
Genetic resilience or resistance in poultry against avian influenza virus: mirage or reality?J Virol. 2025 Jul 22;99(7):e0082025. doi: 10.1128/jvi.00820-25. Epub 2025 Jun 30. J Virol. 2025. PMID: 40586559 Free PMC article. Review.
-
ADAMTS18-fibronectin interaction regulates the morphology of liver sinusoidal endothelial cells.iScience. 2024 Jun 14;27(7):110273. doi: 10.1016/j.isci.2024.110273. eCollection 2024 Jul 19. iScience. 2024. PMID: 39040056 Free PMC article.
-
The Role of Solute Carrier Family Transporters in Hepatic Steatosis and Hepatic Fibrosis.J Clin Transl Hepatol. 2025 Mar 28;13(3):233-252. doi: 10.14218/JCTH.2024.00348. Epub 2025 Jan 22. J Clin Transl Hepatol. 2025. PMID: 40078199 Free PMC article. Review.
-
Transcellular, Interorgan, and Host-microbial Communication in Metabolic Homeostasis: Hearing for the First Time What We Once Thought to be Unspoken.Cell Mol Gastroenterol Hepatol. 2025;19(9):101545. doi: 10.1016/j.jcmgh.2025.101545. Epub 2025 Jun 9. Cell Mol Gastroenterol Hepatol. 2025. PMID: 40532271 Free PMC article. No abstract available.
-
Of Sugar and Fat: How Protein Glycosylation in Sinusoidal Cells Controls Lipid Metabolism in Liver.Cell Mol Gastroenterol Hepatol. 2024;17(6):1064-1065. doi: 10.1016/j.jcmgh.2024.03.010. Epub 2024 Apr 5. Cell Mol Gastroenterol Hepatol. 2024. PMID: 38583483 Free PMC article. No abstract available.
References
-
- Ober E.A., Lemaigre F.P. Development of the liver: insights into organ and tissue morphogenesis. J Hepatol. 2018;68:1049–1062. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
