

Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of protein homeostasis disruptions
- PMID: 38467653
- PMCID: PMC10928085
- DOI: 10.1038/s41467-024-46600-5
Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of protein homeostasis disruptions
Abstract
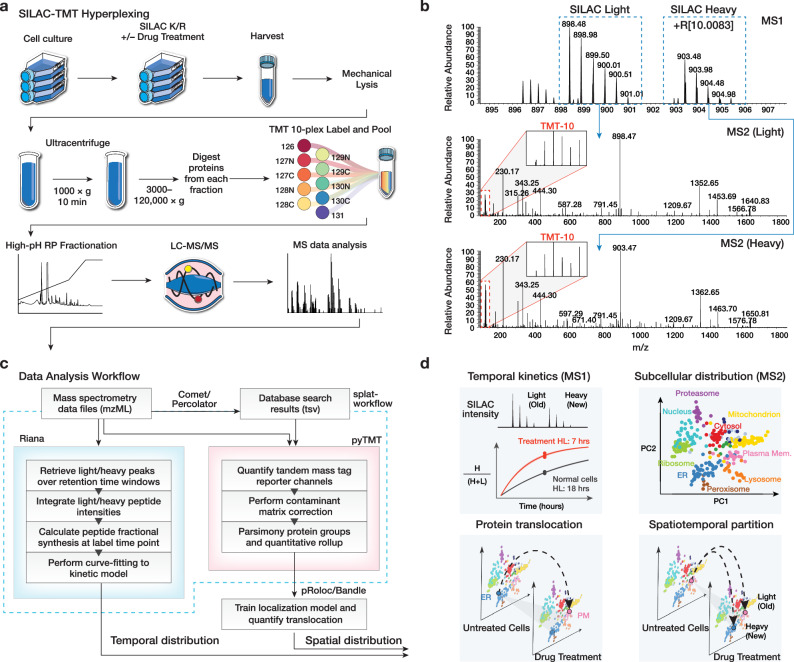
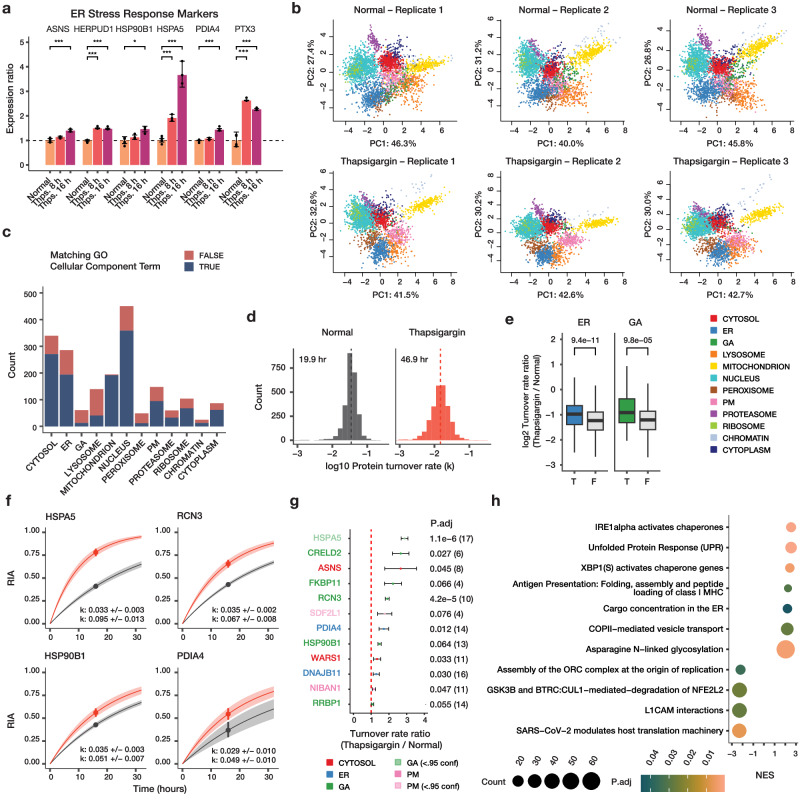
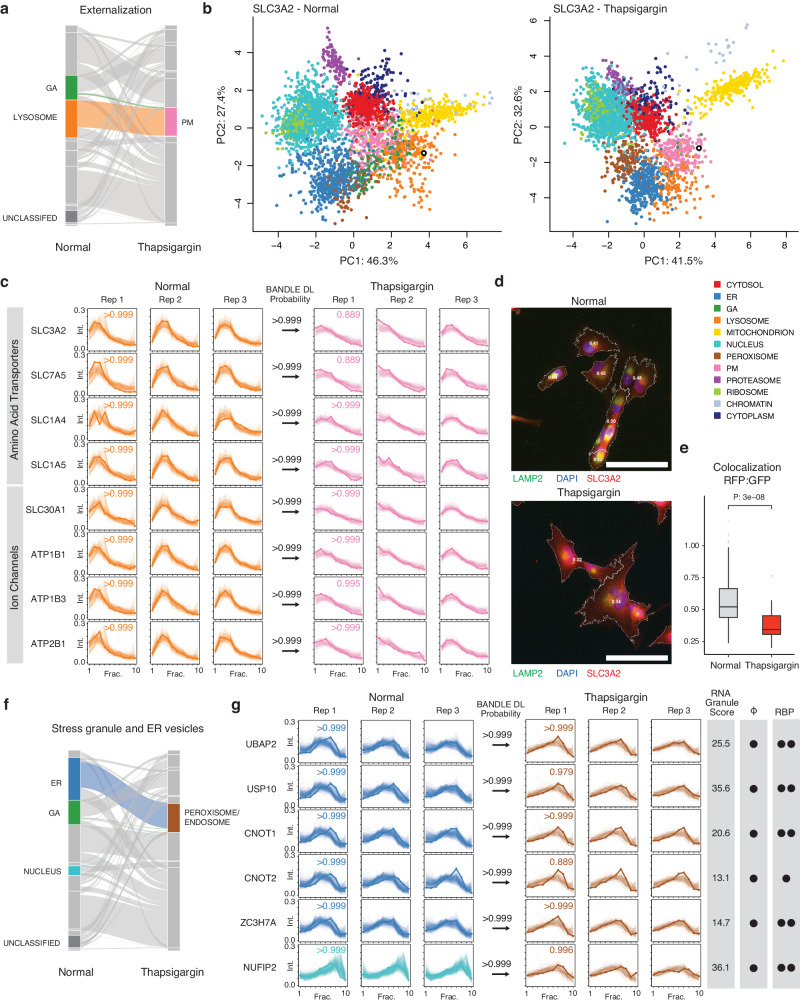
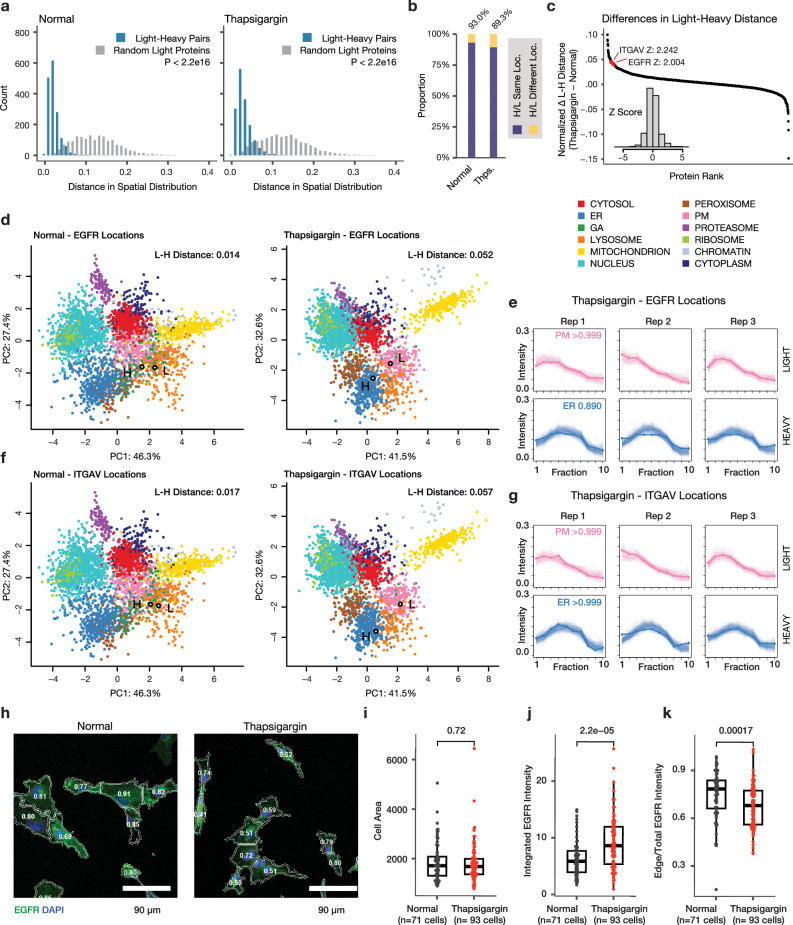
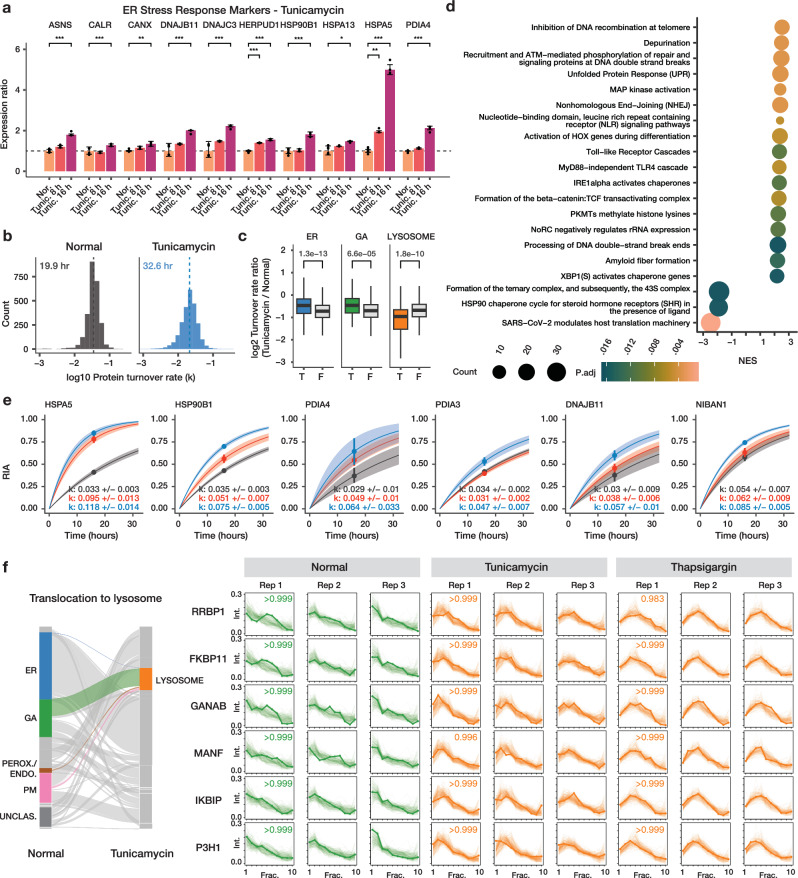
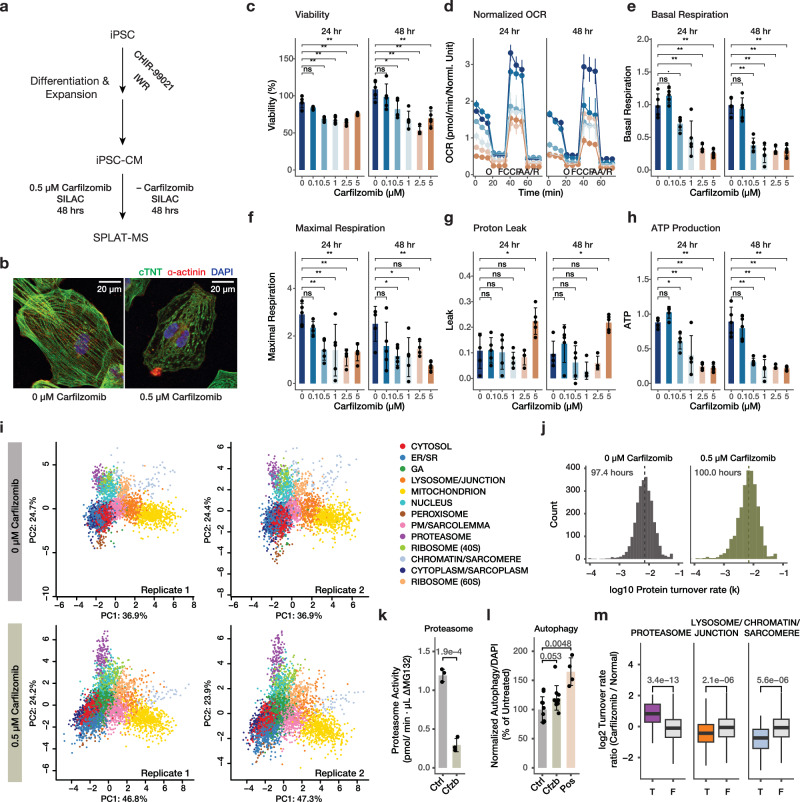
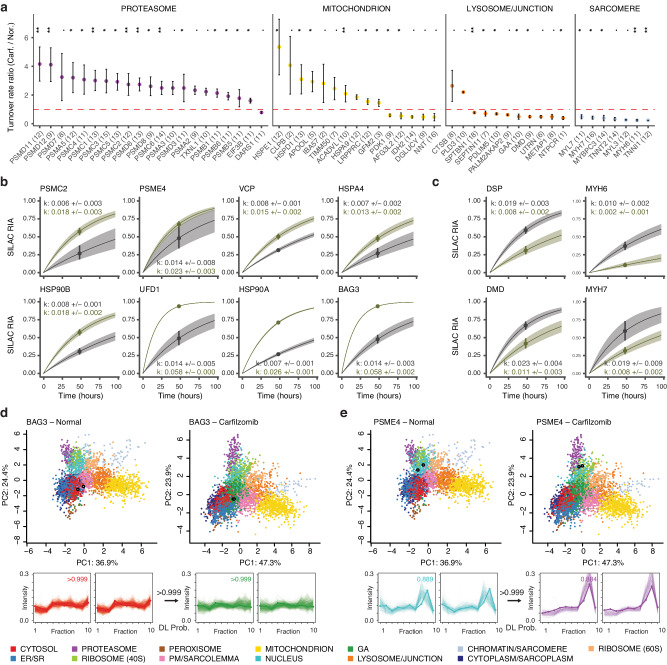
The spatial and temporal distributions of proteins are critical to protein function, but cannot be directly assessed by measuring protein bundance. Here we describe a mass spectrometry-based proteomics strategy, Simultaneous Proteome Localization and Turnover (SPLAT), to measure concurrently protein turnover rates and subcellular localization in the same experiment. Applying the method, we find that unfolded protein response (UPR) has different effects on protein turnover dependent on their subcellular location in human AC16 cells, with proteome-wide slowdown but acceleration among stress response proteins in the ER and Golgi. In parallel, UPR triggers broad differential localization of proteins including RNA-binding proteins and amino acid transporters. Moreover, we observe newly synthesized proteins including EGFR that show a differential localization under stress than the existing protein pools, reminiscent of protein trafficking disruptions. We next applied SPLAT to an induced pluripotent stem cell derived cardiomyocyte (iPSC-CM) model of cancer drug cardiotoxicity upon treatment with the proteasome inhibitor carfilzomib. Paradoxically, carfilzomib has little effect on global average protein half-life, but may instead selectively disrupt sarcomere protein homeostasis. This study provides a view into the interactions of protein spatial and temporal dynamics and demonstrates a method to examine protein homeostasis regulations in stress and drug response.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of protein homeostasis disruptions.bioRxiv [Preprint]. 2024 Jan 17:2023.01.04.521821. doi: 10.1101/2023.01.04.521821. bioRxiv. 2024. Update in: Nat Commun. 2024 Mar 11;15(1):2207. doi: 10.1038/s41467-024-46600-5. PMID: 36711879 Free PMC article. Updated. Preprint.
Similar articles
-
Simultaneous proteome localization and turnover analysis reveals spatiotemporal features of protein homeostasis disruptions.bioRxiv [Preprint]. 2024 Jan 17:2023.01.04.521821. doi: 10.1101/2023.01.04.521821. bioRxiv. 2024. Update in: Nat Commun. 2024 Mar 11;15(1):2207. doi: 10.1038/s41467-024-46600-5. PMID: 36711879 Free PMC article. Updated. Preprint.
-
A quantitative spatial proteomics analysis of proteome turnover in human cells.Mol Cell Proteomics. 2012 Mar;11(3):M111.011429. doi: 10.1074/mcp.M111.011429. Epub 2011 Sep 21. Mol Cell Proteomics. 2012. PMID: 21937730 Free PMC article.
-
Rescue of F508del-CFTR by RXR motif inactivation triggers proteome modulation associated with the unfolded protein response.Biochim Biophys Acta. 2010 Apr;1804(4):856-65. doi: 10.1016/j.bbapap.2009.12.013. Epub 2010 Jan 4. Biochim Biophys Acta. 2010. PMID: 20044041
-
Mechanisms of protein homeostasis (proteostasis) maintain stem cell identity in mammalian pluripotent stem cells.Cell Mol Life Sci. 2018 Jan;75(2):275-290. doi: 10.1007/s00018-017-2602-1. Epub 2017 Jul 26. Cell Mol Life Sci. 2018. PMID: 28748323 Free PMC article. Review.
-
Revealing functional insights into ER proteostasis through proteomics and interactomics.Exp Cell Res. 2021 Feb 1;399(1):112417. doi: 10.1016/j.yexcr.2020.112417. Epub 2020 Dec 8. Exp Cell Res. 2021. PMID: 33301765 Free PMC article. Review.
Cited by
-
A Ratiometric Catalog of Protein Isoform Shifts in the Cardiac Fetal Gene Program.bioRxiv [Preprint]. 2025 May 31:2024.04.09.588716. doi: 10.1101/2024.04.09.588716. bioRxiv. 2025. Update in: JCI Insight. 2025 Aug 7:e184309. doi: 10.1172/jci.insight.184309. PMID: 38645170 Free PMC article. Updated. Preprint.
-
Application of Spatial Omics in the Cardiovascular System.Research (Wash D C). 2025 Mar 8;8:0628. doi: 10.34133/research.0628. eCollection 2025. Research (Wash D C). 2025. PMID: 40062231 Free PMC article. Review.
-
Proteomics and Machine Learning-Based Approach to Decipher Subcellular Proteome of Mouse Heart.Mol Cell Proteomics. 2025 Apr;24(4):100952. doi: 10.1016/j.mcpro.2025.100952. Epub 2025 Mar 18. Mol Cell Proteomics. 2025. PMID: 40113211 Free PMC article.
-
Small Molecule-Induced Alterations of Protein Polyubiquitination Revealed by Mass-Spectrometric Ubiquitome Analysis.Angew Chem Int Ed Engl. 2025 Aug 4;64(32):e202508916. doi: 10.1002/anie.202508916. Epub 2025 Jun 26. Angew Chem Int Ed Engl. 2025. PMID: 40444580 Free PMC article.
-
Deuterium labeling enables proteome wide turnover kinetics analysis in cell culture.bioRxiv [Preprint]. 2025 Jan 31:2025.01.30.635596. doi: 10.1101/2025.01.30.635596. bioRxiv. 2025. Update in: Cell Rep Methods. 2025 Jul 21;5(7):101104. doi: 10.1016/j.crmeth.2025.101104. PMID: 39975278 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
