

A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes
- PMID: 38468278
- PMCID: PMC10929077
- DOI: 10.1186/s13059-024-03207-9
A comparison of methods for detecting DNA methylation from long-read sequencing of human genomes
Abstract
Background: Long-read sequencing can enable the detection of base modifications, such as CpG methylation, in single molecules of DNA. The most commonly used methods for long-read sequencing are nanopore developed by Oxford Nanopore Technologies (ONT) and single molecule real-time (SMRT) sequencing developed by Pacific Bioscience (PacBio). In this study, we systematically compare the performance of CpG methylation detection from long-read sequencing.
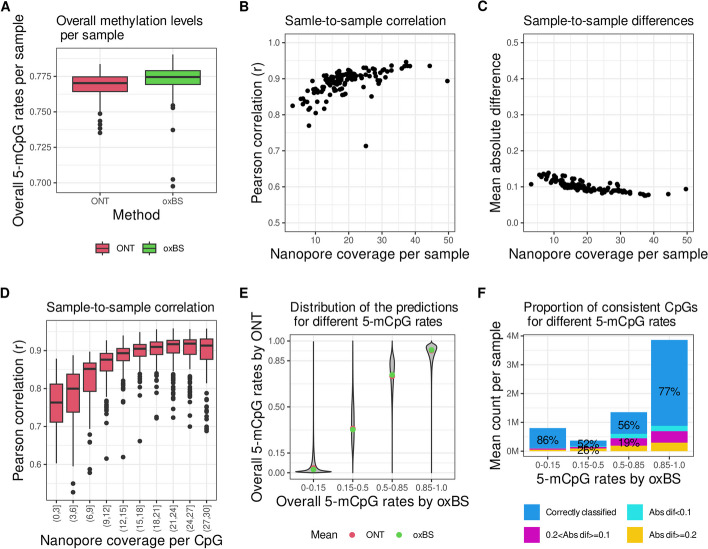
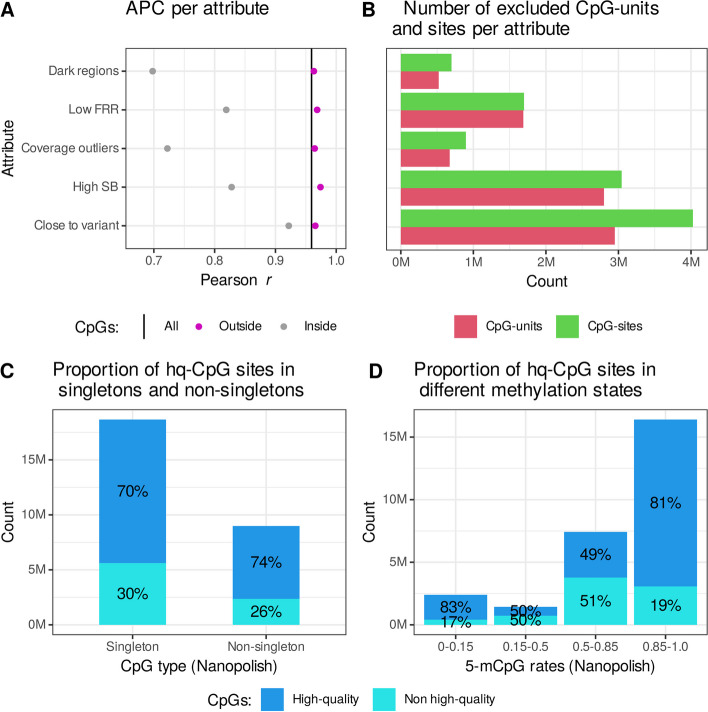
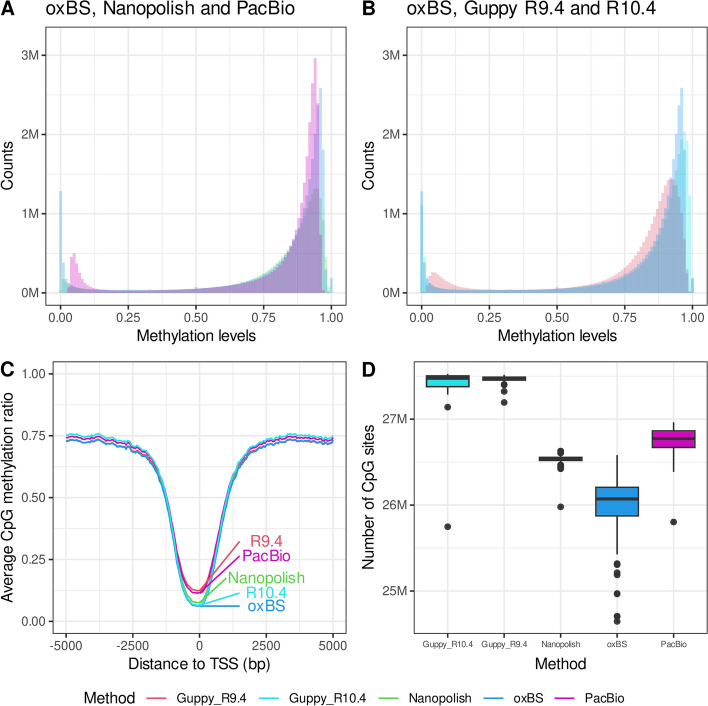
Results: We demonstrate that CpG methylation detection from 7179 nanopore-sequenced DNA samples is highly accurate and consistent with 132 oxidative bisulfite-sequenced (oxBS) samples, isolated from the same blood draws. We introduce quality filters for CpGs that further enhance the accuracy of CpG methylation detection from nanopore-sequenced DNA, while removing at most 30% of CpGs. We evaluate the per-site performance of CpG methylation detection across different genomic features and CpG methylation rates and demonstrate how the latest R10.4 flowcell chemistry and base-calling algorithms improve methylation detection from nanopore sequencing. Additionally, we show how the methylation detection of 50 SMRT-sequenced genomes compares to nanopore sequencing and oxBS.
Conclusions: This study provides the first systematic comparison of CpG methylation detection tools for long-read sequencing methods. We compare two commonly used computational methods for the detection of CpG methylation in a large number of nanopore genomes, including samples sequenced using the latest R10.4 nanopore flowcell chemistry and 50 SMRT sequenced samples. We provide insights into the strengths and limitations of each sequencing method as well as recommendations for standardization and evaluation of tools designed for genome-scale modified base detection using long-read sequencing.
© 2024. The Author(s).
Conflict of interest statement
All authors are employees of deCODE genetics/Amgen, Inc.
Figures
References
-
- Tost J, Gut IG. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol Biol. 2007;373:89–102. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
