

Comprehensive phenotypes of patients with SYNGAP1-related disorder reveals high rates of epilepsy and autism
- PMID: 38470175
- PMCID: PMC12375243
- DOI: 10.1111/epi.17913
Comprehensive phenotypes of patients with SYNGAP1-related disorder reveals high rates of epilepsy and autism
Abstract
Objective: To delineate the comprehensive phenotypic spectrum of SYNGAP1-related disorder in a large patient cohort aggregated through a digital registry.
Methods: We obtained de-identified patient data from an online registry. Data were extracted from uploaded medical records. We reclassified all SYNGAP1 variants using American College of Medical Genetics criteria and included patients with pathogenic/likely pathogenic (P/LP) single nucleotide variants or microdeletions incorporating SYNGAP1. We analyzed neurodevelopmental phenotypes, including epilepsy, intellectual disability (ID), autism spectrum disorder (ASD), behavioral disorders, and gait dysfunction for all patients with respect to variant type and location within the SynGAP1 protein.
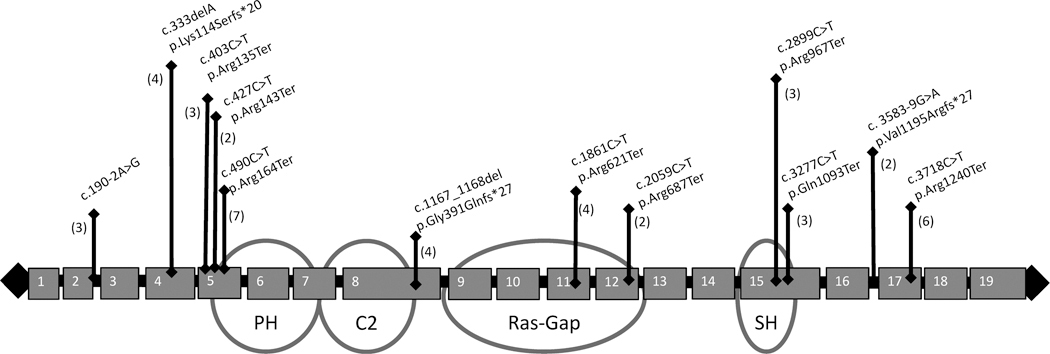
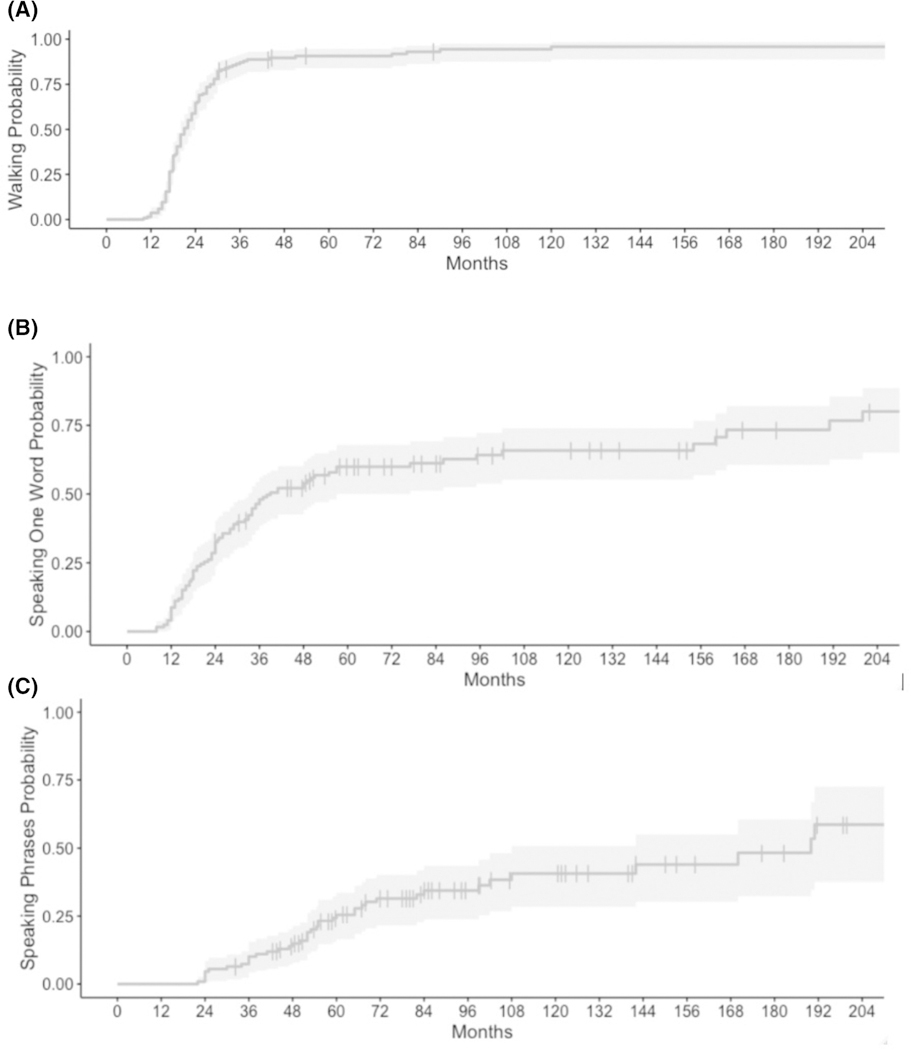
Results: We identified 147 patients (50% male, median age 8 years) with P/LP SYNGAP1 variants from 151 individuals with data available through the database. One hundred nine were truncating variants and 22 were missense. All patients were diagnosed with global developmental delay (GDD) and/or ID, and 123 patients (84%) were diagnosed with epilepsy. Of those with epilepsy, 73% of patients had GDD diagnosed before epilepsy was diagnosed. Other prominent features included autistic traits (n = 100, 68%), behavioral problems (n = 100, 68%), sleep problems (n = 90, 61%), anxiety (n = 35, 24%), ataxia or abnormal gait (n = 69, 47%), sensory problems (n = 32, 22%), and feeding difficulties (n = 69, 47%). Behavioral problems were more likely in those patients diagnosed with anxiety (odds ratio [OR] 3.6, p = .014) and sleep problems (OR 2.41, p = .015) but not necessarily those with autistic traits. Patients with variants in exons 1-4 were more likely to have the ability to speak in phrases vs those with variants in exons 5-19, and epilepsy occurred less frequently in patients with variants in the SH3 binding motif.
Significance: We demonstrate that the data obtained from a digital registry recapitulate earlier but smaller studies of SYNGAP1-related disorder and add additional genotype-phenotype relationships, validating the use of the digital registry. Access to data through digital registries broadens the possibilities for efficient data collection in rare diseases.
Keywords: SYNGAP1; developmental and epileptic encephalopathy; epilepsy with eyelid myoclonia; epilepsy with myoclonic–atonic seizures; genetic generalized epilepsy; intellectual disability.
© 2024 International League Against Epilepsy.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
Elise Brimble is employed by Invitae. Kimberly Wiltrout is a consultant for Stoke Therapeutics. Annapurna Poduri serves, without compensation, on the scientific advisory board for The SynGAP Research Fund. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Figures



Similar articles
-
Behavioural phenotype of SYNGAP1-related intellectual disability.J Intellect Disabil Res. 2024 Sep;68(9):1036-1049. doi: 10.1111/jir.13145. Epub 2024 May 23. J Intellect Disabil Res. 2024. PMID: 38783394
-
Subjective sleep assessment in individuals with SYNGAP1-associated syndrome.J Clin Sleep Med. 2024 Dec 1;20(12):1879-1885. doi: 10.5664/jcsm.11246. J Clin Sleep Med. 2024. PMID: 38958060
-
SYNGAP1-Related Intellectual Disability.2019 Feb 21 [updated 2025 Aug 14]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2019 Feb 21 [updated 2025 Aug 14]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 30789692 Free Books & Documents. Review.
-
MBOAT7 encephalopathy: Characterizing the neurology and epileptology.Epilepsia. 2025 Jul;66(7):2379-2390. doi: 10.1111/epi.18376. Epub 2025 Mar 21. Epilepsia. 2025. PMID: 40116760 Free PMC article.
-
Prognosis of adults and children following a first unprovoked seizure.Cochrane Database Syst Rev. 2023 Jan 23;1(1):CD013847. doi: 10.1002/14651858.CD013847.pub2. Cochrane Database Syst Rev. 2023. PMID: 36688481 Free PMC article.
Cited by
-
Distinct features of EEG microstates in autism spectrum disorder revealed by meta-analysis: the contribution of individual age to heterogeneity across studies.Front Psychiatry. 2025 Apr 22;16:1531694. doi: 10.3389/fpsyt.2025.1531694. eCollection 2025. Front Psychiatry. 2025. PMID: 40330653 Free PMC article.
-
Presymptomatic Biological, Structural, and Functional Diagnostic Biomarkers of Autism Spectrum Disorder.J Neurochem. 2025 May;169(5):e70088. doi: 10.1111/jnc.70088. J Neurochem. 2025. PMID: 40390287 Free PMC article.
-
Analysis of retrospective natural history data collected from patients with SYNGAP1-related disorders: a preliminary examination of the Citizen database.Orphanet J Rare Dis. 2025 Jul 27;20(1):379. doi: 10.1186/s13023-025-03918-7. Orphanet J Rare Dis. 2025. PMID: 40717090 Free PMC article.
-
Atomistic simulations reveal impacts of missense mutations on the structure and function of SynGAP1.Brief Bioinform. 2024 Sep 23;25(6):bbae458. doi: 10.1093/bib/bbae458. Brief Bioinform. 2024. PMID: 39311700 Free PMC article.
-
SYNGAP1 Syndrome and the Brain Gene Registry.Genes (Basel). 2025 Mar 30;16(4):405. doi: 10.3390/genes16040405. Genes (Basel). 2025. PMID: 40282364 Free PMC article. Review.
References
-
- Agarwal M, Johnston MV, Stafstrom CE. SYNGAP1 mutations: clinical, genetic, and pathophysiological features. Int J Dev Neurosci. 2019;78:65–76. - PubMed
-
- Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–91. - PubMed
-
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20:895–904. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
