

Comprehensive transcriptome analysis reveals altered mRNA splicing and post-transcriptional changes in the aged mouse brain
- PMID: 38471806
- PMCID: PMC11014377
- DOI: 10.1093/nar/gkae172
Comprehensive transcriptome analysis reveals altered mRNA splicing and post-transcriptional changes in the aged mouse brain
Abstract
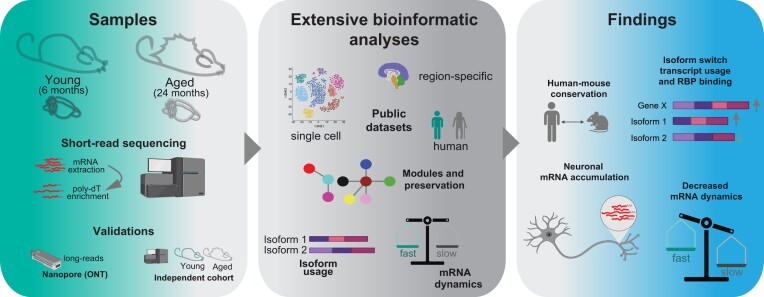
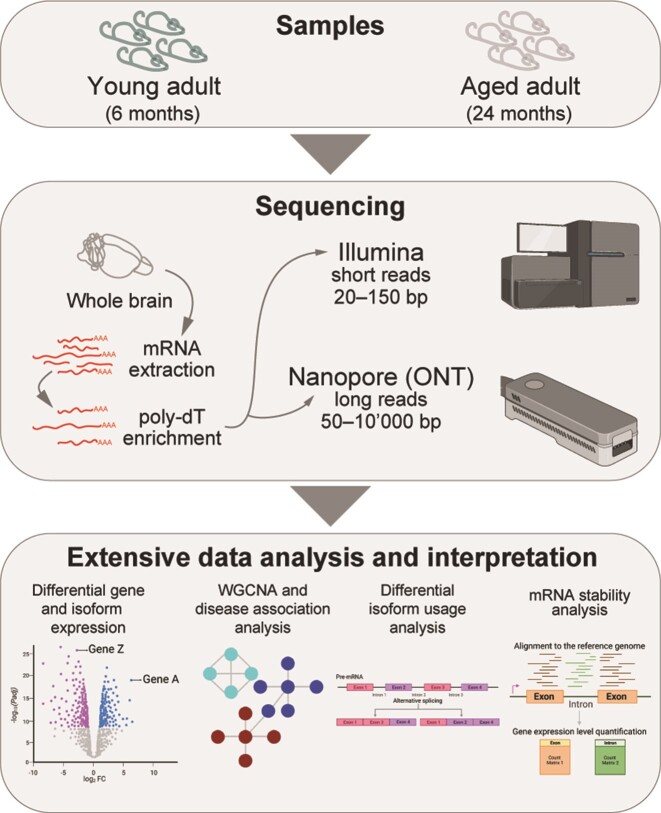
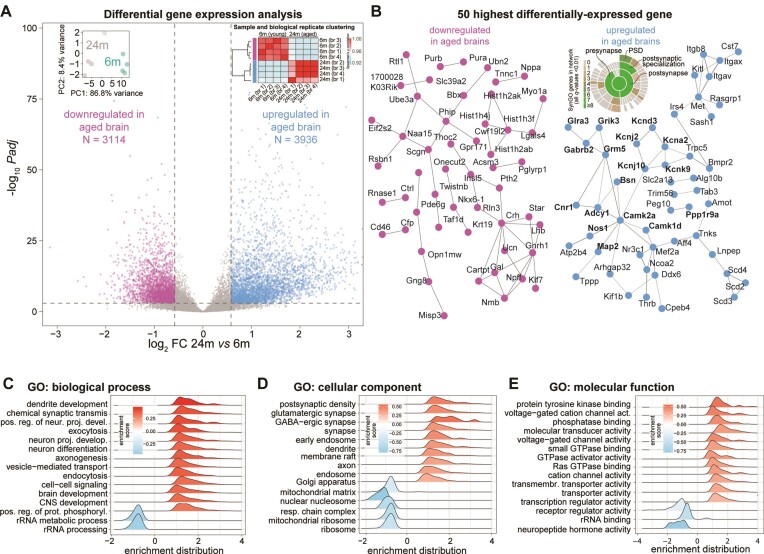
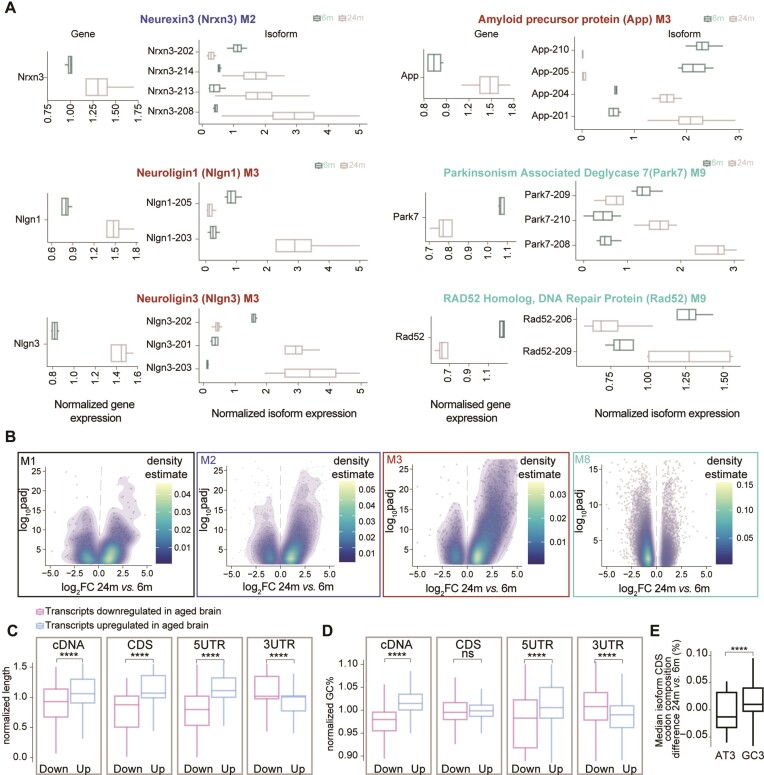
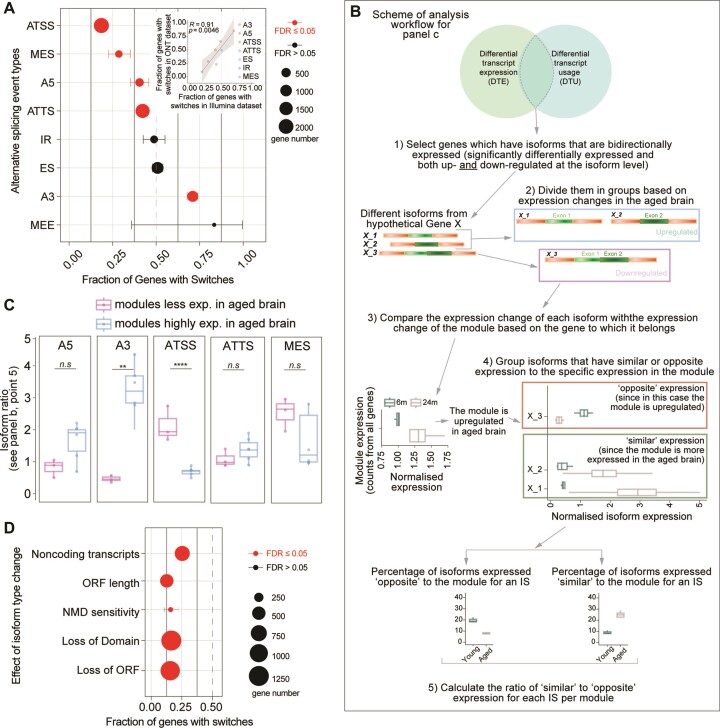
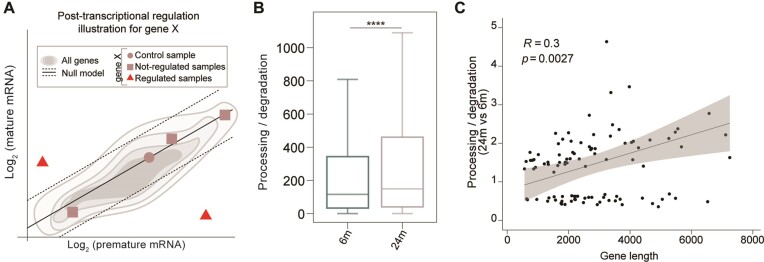
A comprehensive understanding of molecular changes during brain aging is essential to mitigate cognitive decline and delay neurodegenerative diseases. The interpretation of mRNA alterations during brain aging is influenced by the health and age of the animal cohorts studied. Here, we carefully consider these factors and provide an in-depth investigation of mRNA splicing and dynamics in the aging mouse brain, combining short- and long-read sequencing technologies with extensive bioinformatic analyses. Our findings encompass a spectrum of age-related changes, including differences in isoform usage, decreased mRNA dynamics and a module showing increased expression of neuronal genes. Notably, our results indicate a reduced abundance of mRNA isoforms leading to nonsense-mediated RNA decay and suggest a regulatory role for RNA-binding proteins, indicating that their regulation may be altered leading to the reshaping of the aged brain transcriptome. Collectively, our study highlights the importance of studying mRNA splicing events during brain aging.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
