

Viral reprogramming of host transcription initiation
- PMID: 38471819
- PMCID: PMC11109974
- DOI: 10.1093/nar/gkae175
Viral reprogramming of host transcription initiation
Abstract
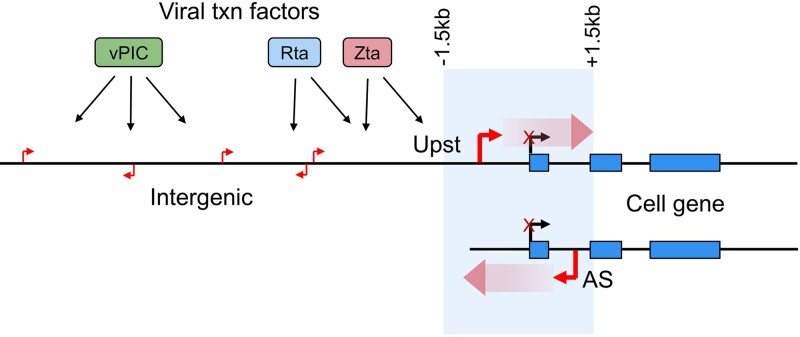
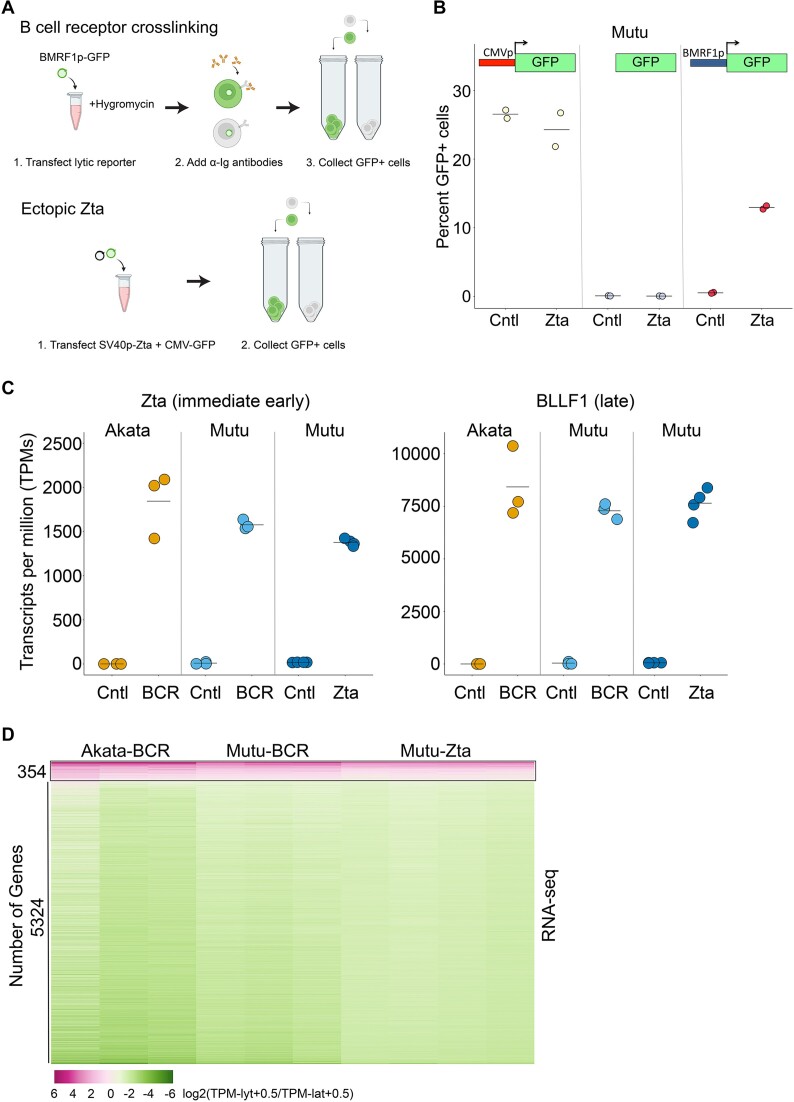
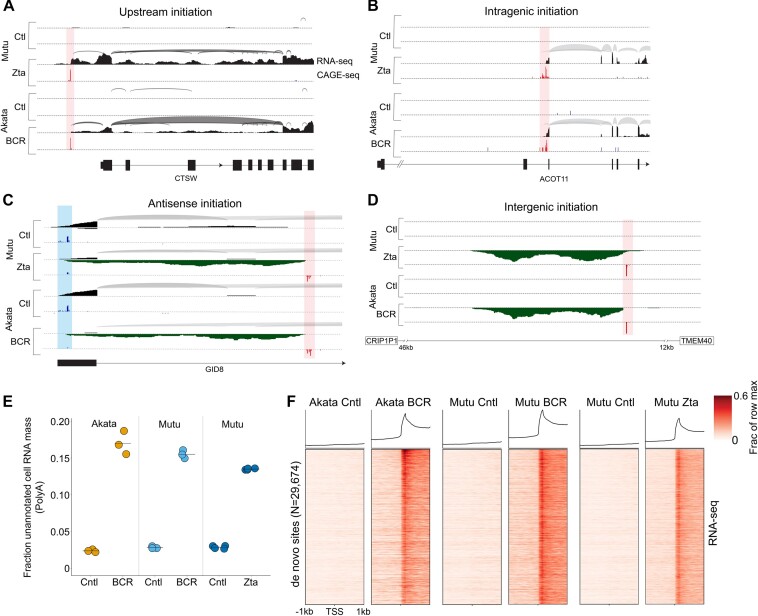
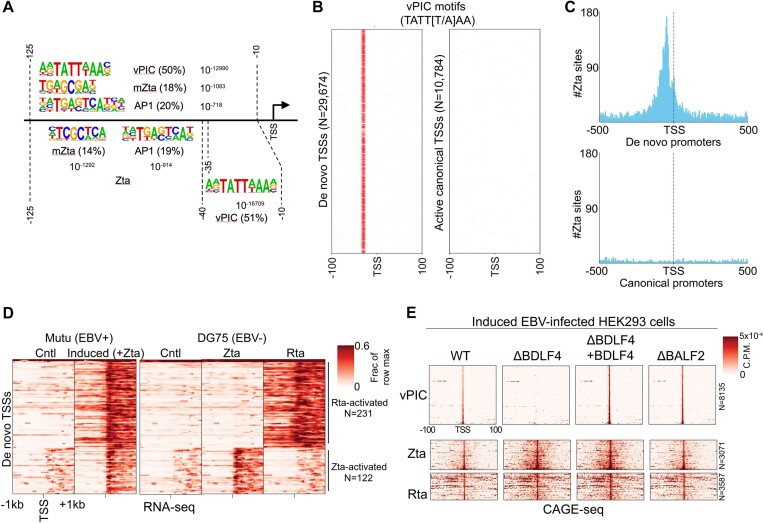
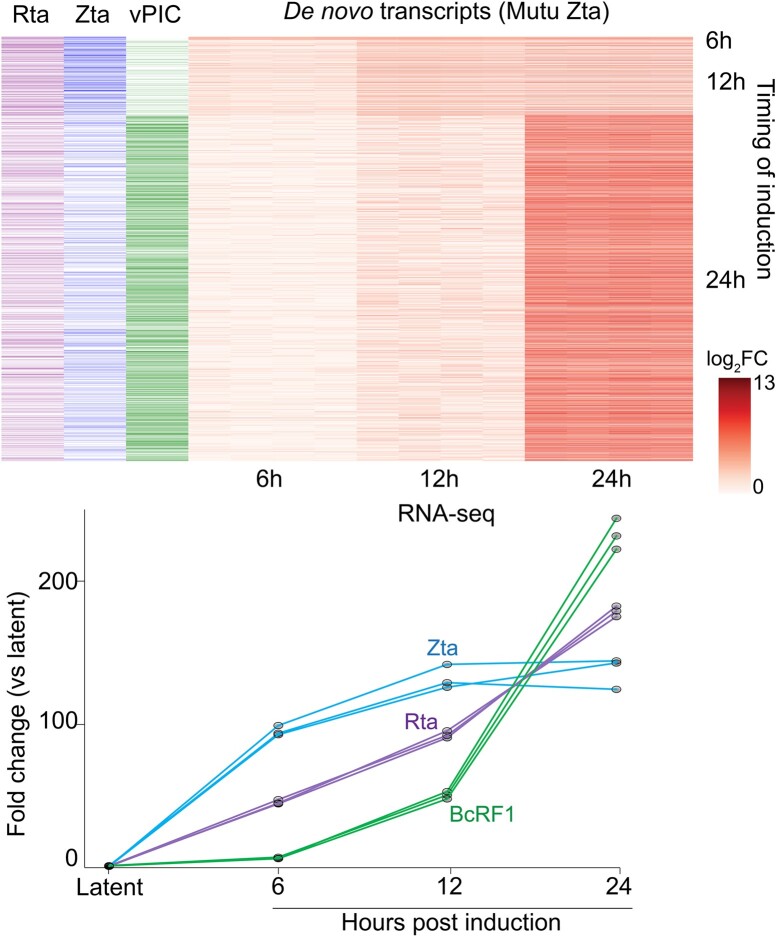
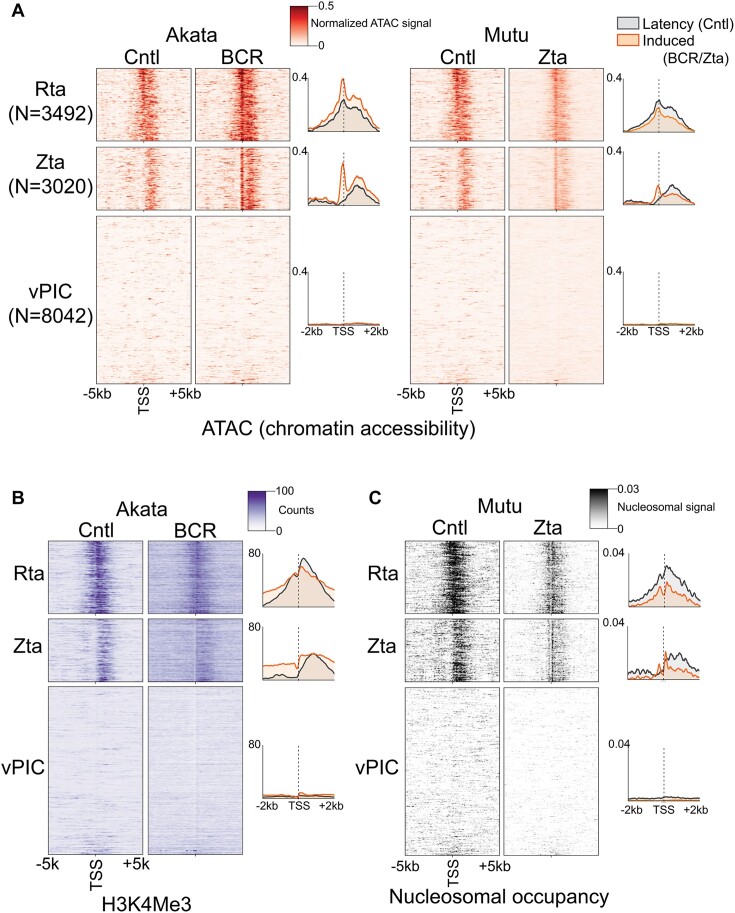
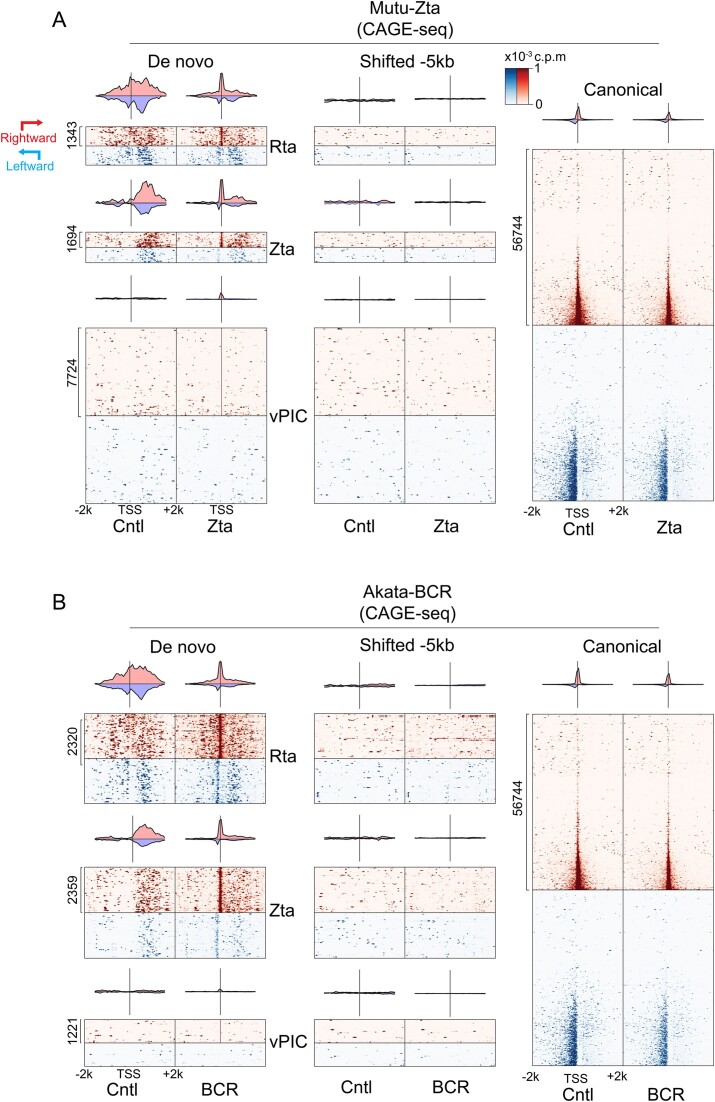
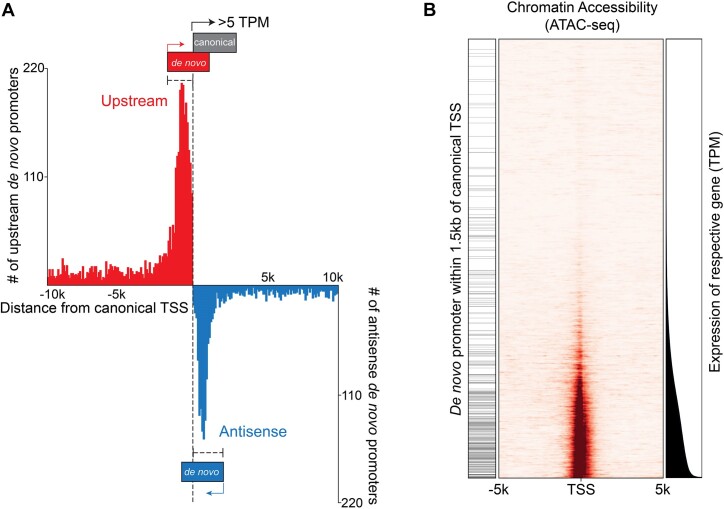
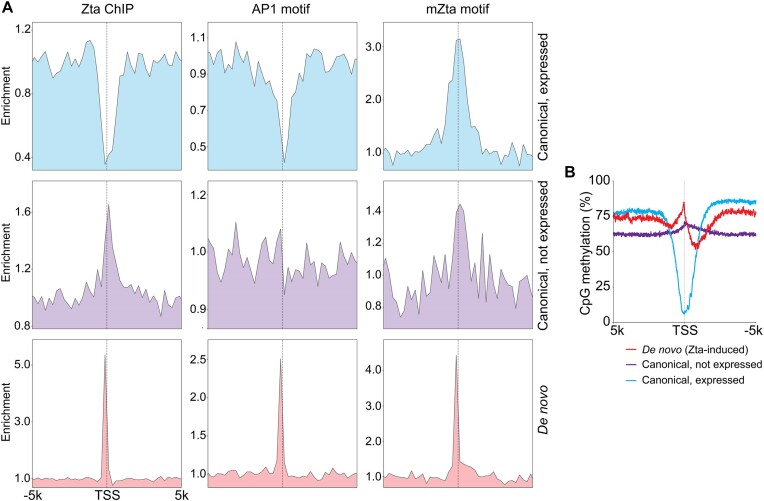
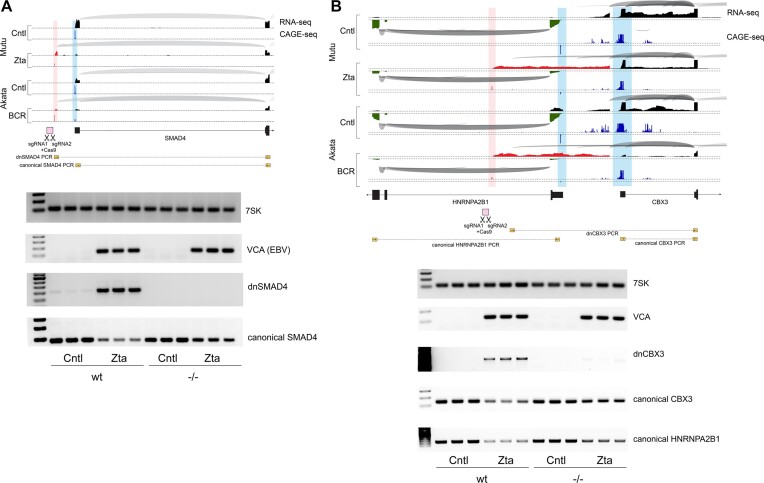
Viruses are master remodelers of the host cell environment in support of infection and virus production. For example, viruses typically regulate cell gene expression through modulating canonical cell promoter activity. Here, we show that Epstein Barr virus (EBV) replication causes 'de novo' transcription initiation at 29674 new transcription start sites throughout the cell genome. De novo transcription initiation is facilitated in part by the unique properties of the viral pre-initiation complex (vPIC) that binds a TATT[T/A]AA, TATA box-like sequence and activates transcription with minimal support by additional transcription factors. Other de novo promoters are driven by the viral transcription factors, Zta and Rta and are influenced by directional proximity to existing canonical cell promoters, a configuration that fosters transcription through existing promoters and transcriptional interference. These studies reveal a new way that viruses interact with the host transcriptome to inhibit host gene expression and they shed light on primal features driving eukaryotic promoter function.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Farrell P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. 2019; 14:29–53. - PubMed
-
- Bjornevik K., Cortese M., Healy B.C., Kuhle J., Mina M.J., Leng Y., Elledge S.J., Niebuhr D.W., Scher A.I., Munger K.L. et al. . Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022; 375:296–301. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
