

Impact of Complex Apoptotic Signaling Pathways on Cancer Cell Sensitivity to Therapy
- PMID: 38473345
- PMCID: PMC10930821
- DOI: 10.3390/cancers16050984
Impact of Complex Apoptotic Signaling Pathways on Cancer Cell Sensitivity to Therapy
Abstract
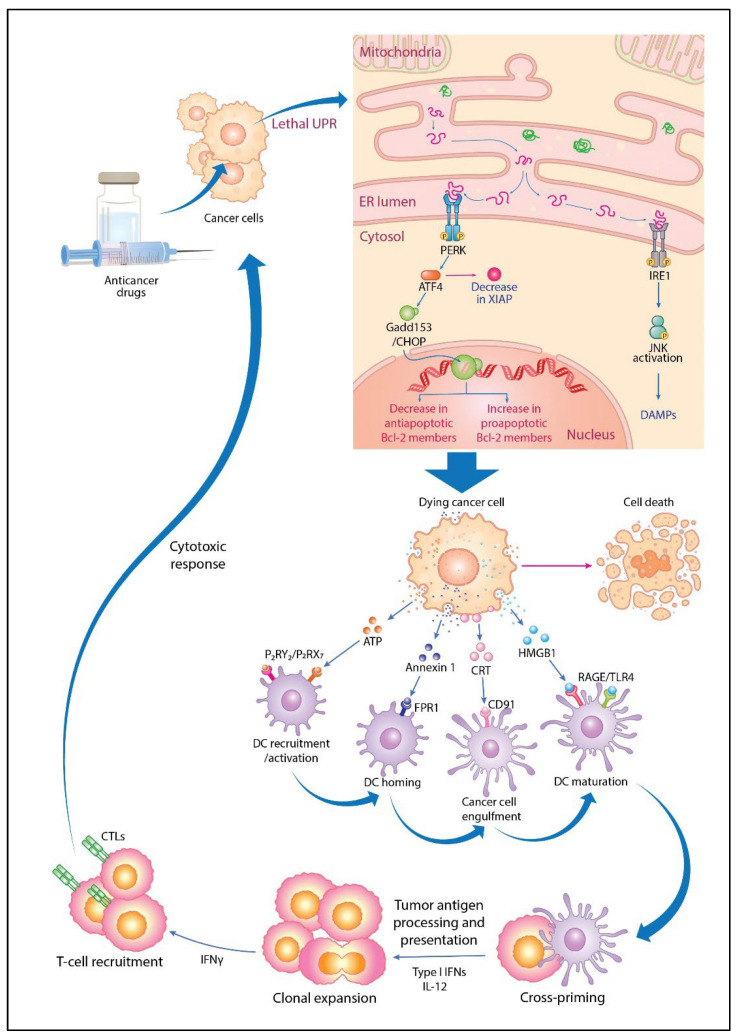
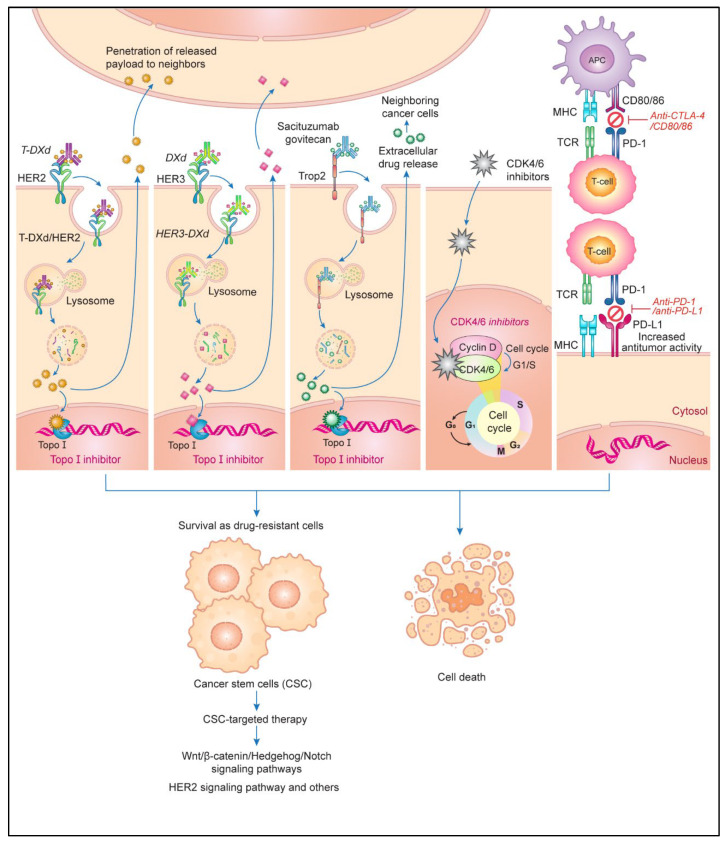
Anticancer drugs induce apoptotic and non-apoptotic cell death in various cancer types. The signaling pathways for anticancer drug-induced apoptotic cell death have been shown to differ between drug-sensitive and drug-resistant cells. In atypical multidrug-resistant leukemia cells, the c-Jun/activator protein 1 (AP-1)/p53 signaling pathway leading to apoptotic death is altered. Cancer cells treated with anticancer drugs undergo c-Jun/AP-1-mediated apoptotic death and are involved in c-Jun N-terminal kinase activation and growth arrest- and DNA damage-inducible gene 153 (Gadd153)/CCAAT/enhancer-binding protein homologous protein pathway induction, regardless of the p53 genotype. Gadd153 induction is associated with mitochondrial membrane permeabilization after anticancer drug treatment and involves a coupled endoplasmic reticulum stress response. The induction of apoptosis by anticancer drugs is mediated by the intrinsic pathway (cytochrome c, Cyt c) and subsequent activation of the caspase cascade via proapoptotic genes (e.g., Bax and Bcl-xS) and their interactions. Anticancer drug-induced apoptosis involves caspase-dependent and caspase-independent pathways and occurs via intrinsic and extrinsic pathways. The targeting of antiapoptotic genes such as Bcl-2 enhances anticancer drug efficacy. The modulation of apoptotic signaling by Bcl-xS transduction increases the sensitivity of multidrug resistance-related protein-overexpressing epidermoid carcinoma cells to anticancer drugs. The significance of autophagy in cancer therapy remains to be elucidated. In this review, we summarize current knowledge of cancer cell death-related signaling pathways and their alterations during anticancer drug treatment and discuss potential strategies to enhance treatment efficacy.
Keywords: anticancer drug; antitumor immunity; cancer cell; cell death; signaling pathway.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
Similar articles
-
Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions.Autophagy. 2014;10(11):1921-36. doi: 10.4161/15548627.2014.981790. Autophagy. 2014. PMID: 25470234 Free PMC article.
-
Cancer and Apoptosis.Methods Mol Biol. 2022;2543:191-210. doi: 10.1007/978-1-0716-2553-8_16. Methods Mol Biol. 2022. PMID: 36087269
-
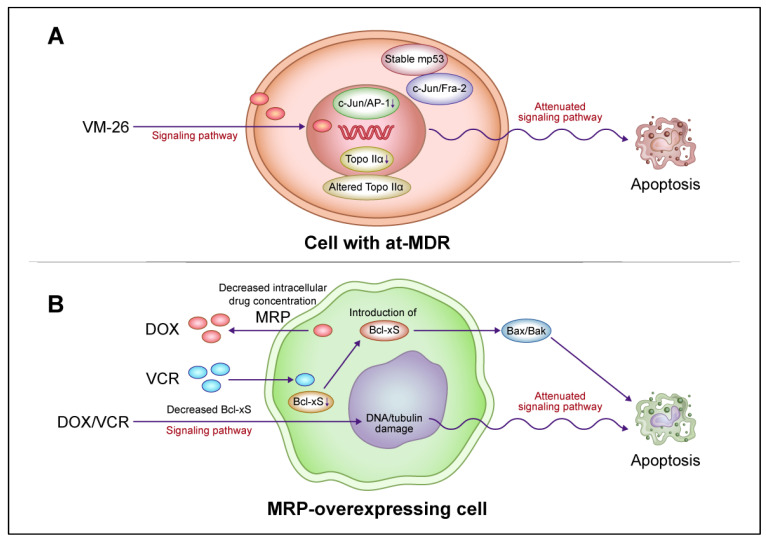
Overcoming of multidrug resistance by introducing the apoptosis gene, bcl-Xs, into MRP-overexpressing drug resistant cells.Int J Oncol. 2000 May;16(5):959-69. doi: 10.3892/ijo.16.5.959. Int J Oncol. 2000. PMID: 10762632
-
Current status of the molecular mechanisms of anticancer drug-induced apoptosis. The contribution of molecular-level analysis to cancer chemotherapy.Cancer Chemother Pharmacol. 2002 Nov;50(5):343-52. doi: 10.1007/s00280-002-0522-7. Epub 2002 Oct 3. Cancer Chemother Pharmacol. 2002. PMID: 12439591 Review.
-
Inducing cancer cell death by targeting transcription factors.Anticancer Drugs. 2003 Jan;14(1):3-11. doi: 10.1097/00001813-200301000-00002. Anticancer Drugs. 2003. PMID: 12544253 Review.
Cited by
-
Anastasis and Other Apoptosis-Related Prosurvival Pathways Call for a Paradigm Shift in Oncology: Significance of Deintensification in Treating Solid Tumors.Int J Mol Sci. 2025 Feb 22;26(5):1881. doi: 10.3390/ijms26051881. Int J Mol Sci. 2025. PMID: 40076508 Free PMC article. Review.
-
Cell death in acute lung injury: caspase-regulated apoptosis, pyroptosis, necroptosis, and PANoptosis.Front Pharmacol. 2025 Mar 21;16:1559659. doi: 10.3389/fphar.2025.1559659. eCollection 2025. Front Pharmacol. 2025. PMID: 40191423 Free PMC article. Review.
-
From copper homeostasis to cuproptosis: a new perspective on CNS immune regulation and neurodegenerative diseases.Front Neurol. 2025 May 29;16:1581045. doi: 10.3389/fneur.2025.1581045. eCollection 2025. Front Neurol. 2025. PMID: 40510202 Free PMC article. Review.
-
Amitotic Cell Division, Malignancy, and Resistance to Anticancer Agents: A Tribute to Drs. Walen and Rajaraman.Cancers (Basel). 2024 Sep 8;16(17):3106. doi: 10.3390/cancers16173106. Cancers (Basel). 2024. PMID: 39272964 Free PMC article.
-
The Proapoptotic Effect of MB-653 Is Associated with the Modulation of Metastasis and Invasiveness-Related Signalling Pathways in Human Colorectal Cancer Cells.Biomolecules. 2025 Jan 6;15(1):72. doi: 10.3390/biom15010072. Biomolecules. 2025. PMID: 39858466 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
