

A Comprehensive Retrospective Study on the Mechanisms of Cyclic Mechanical Stretch-Induced Vascular Smooth Muscle Cell Death Underlying Aortic Dissection and Potential Therapeutics for Preventing Acute Aortic Aneurysm and Associated Ruptures
- PMID: 38473793
- PMCID: PMC10931695
- DOI: 10.3390/ijms25052544
A Comprehensive Retrospective Study on the Mechanisms of Cyclic Mechanical Stretch-Induced Vascular Smooth Muscle Cell Death Underlying Aortic Dissection and Potential Therapeutics for Preventing Acute Aortic Aneurysm and Associated Ruptures
Abstract
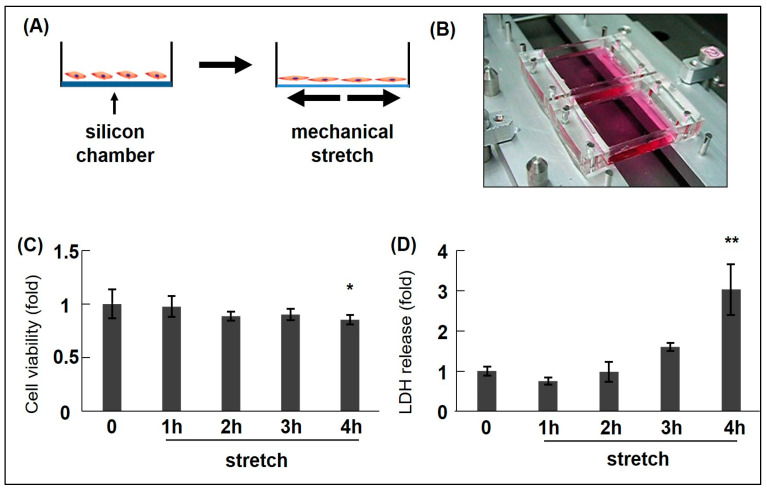
Acute aortic dissection (AAD) and associated ruptures are the leading causes of death in cardiovascular diseases (CVDs). Hypertension is a prime risk factor for AAD. However, the molecular mechanisms underlying AAD remain poorly understood. We previously reported that cyclic mechanical stretch (CMS) leads to the death of rat aortic smooth muscle cells (RASMCs). This review focuses on the mechanisms of CMS-induced vascular smooth muscle cell (VSMC) death. Moreover, we have also discussed the potential therapeutics for preventing AAD and aneurysm ruptures.
Keywords: aortic dissection; cell death; chemokines; hypertension; inducible nitric oxide synthases; mechanical stretch; vascular smooth muscle cell.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
