

The Killer's Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer
- PMID: 38473997
- PMCID: PMC10931665
- DOI: 10.3390/ijms25052750
The Killer's Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer
Abstract
Inflammation is a key contributor to both the initiation and progression of tumors, and it can be triggered by genetic instability within tumors, as well as by lifestyle and dietary factors. The inflammatory response plays a critical role in the genetic and epigenetic reprogramming of tumor cells, as well as in the cells that comprise the tumor microenvironment. Cells in the microenvironment acquire a phenotype that promotes immune evasion, progression, and metastasis. We will review the mechanisms and pathways involved in the interaction between tumors, inflammation, and nutrition, the limitations of current therapies, and discuss potential future therapeutic approaches.
Keywords: DNA repair; cancer; epigenetic; inflammation; nutrition.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
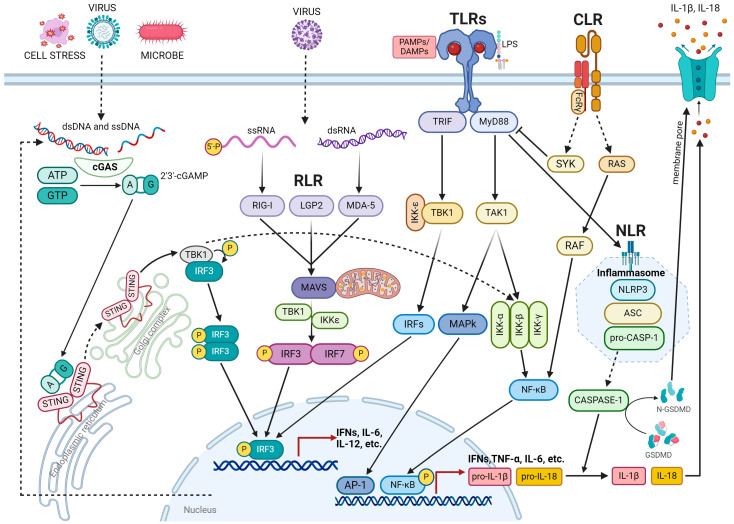
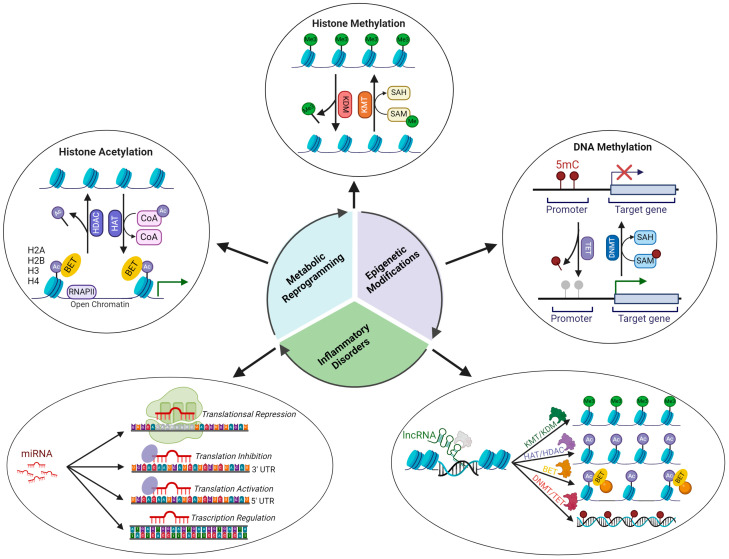
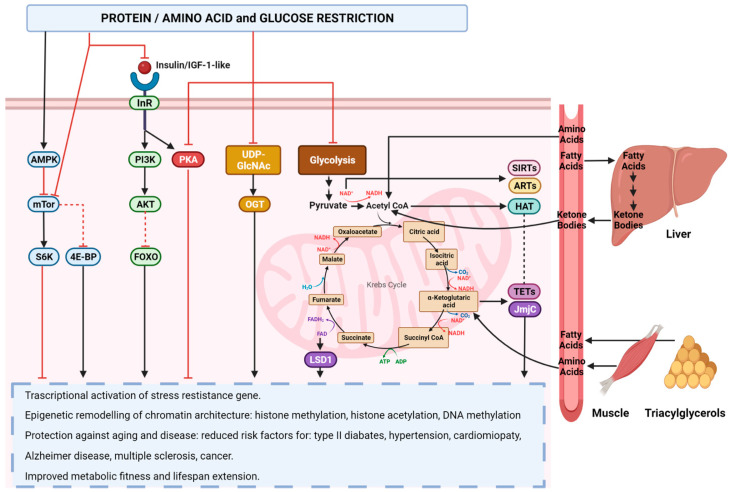
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
