

Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins
- PMID: 38474037
- PMCID: PMC10932171
- DOI: 10.3390/ijms25052790
Protein Kinase D Plays a Crucial Role in Maintaining Cardiac Homeostasis by Regulating Post-Translational Modifications of Myofilament Proteins
Abstract
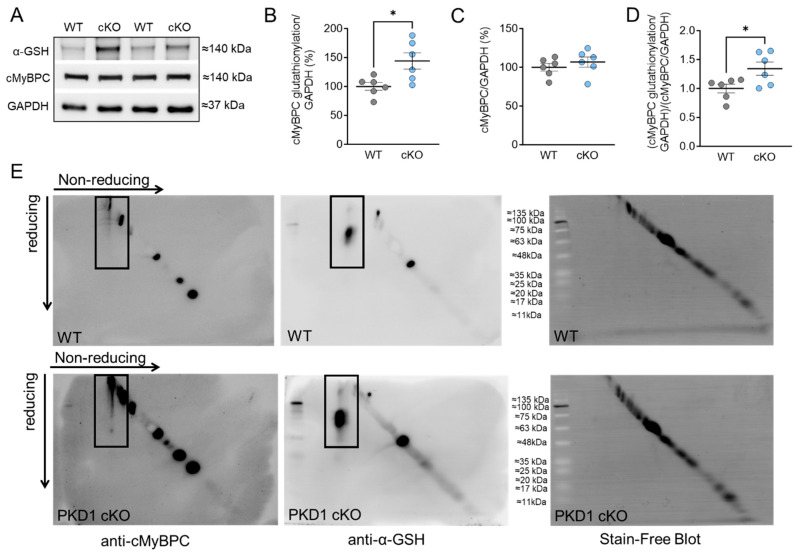
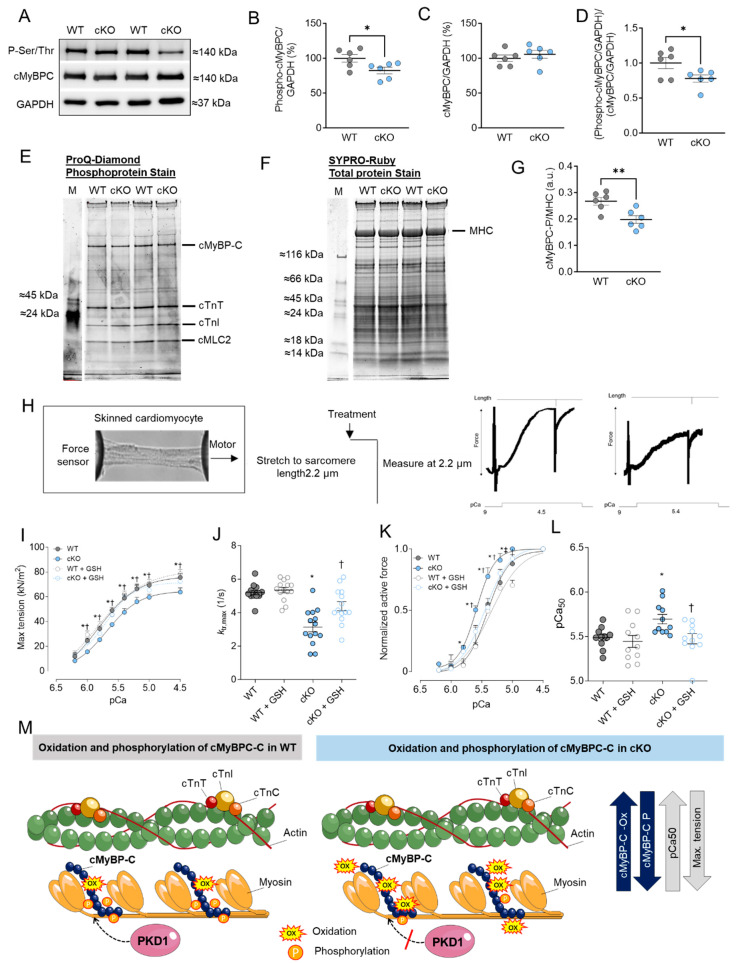
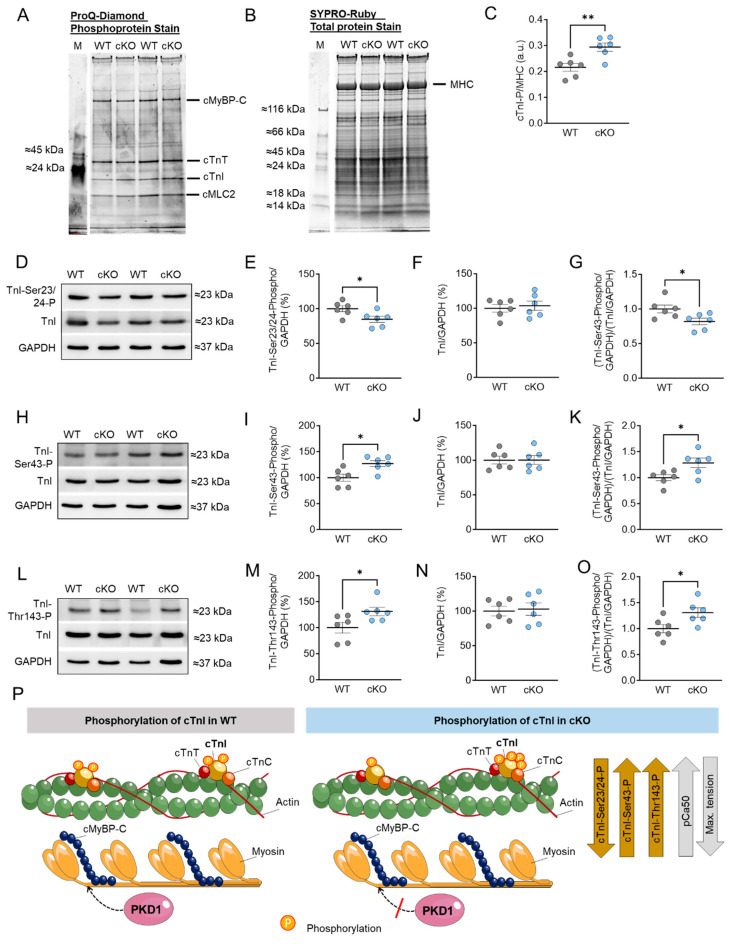
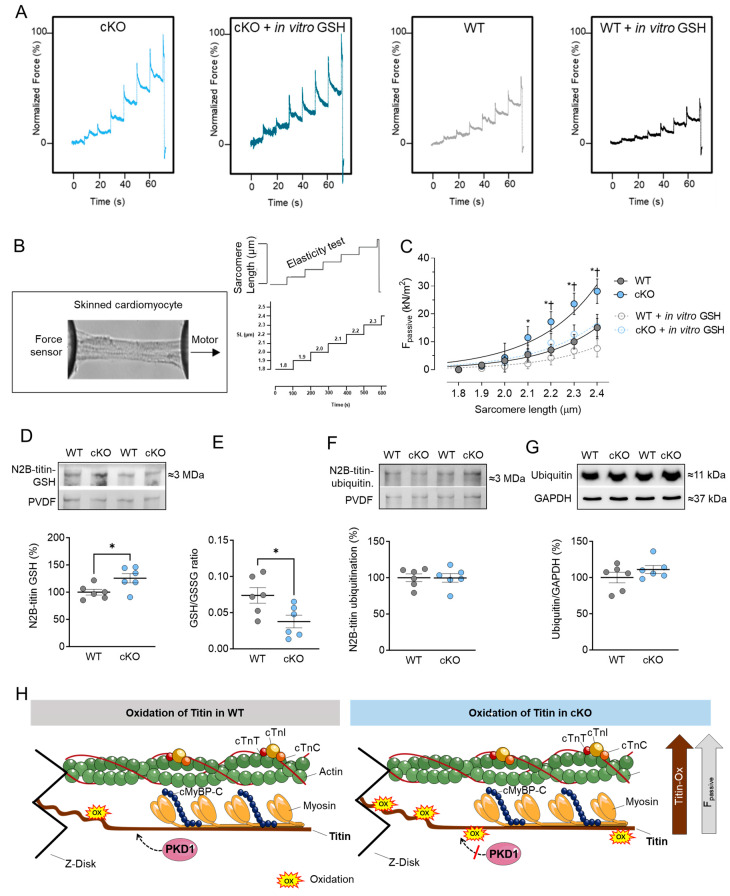
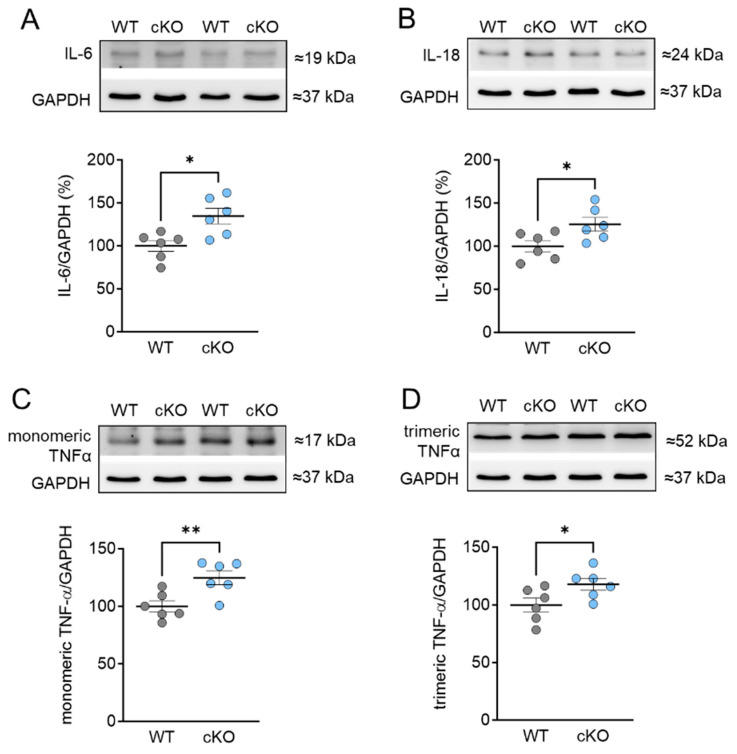
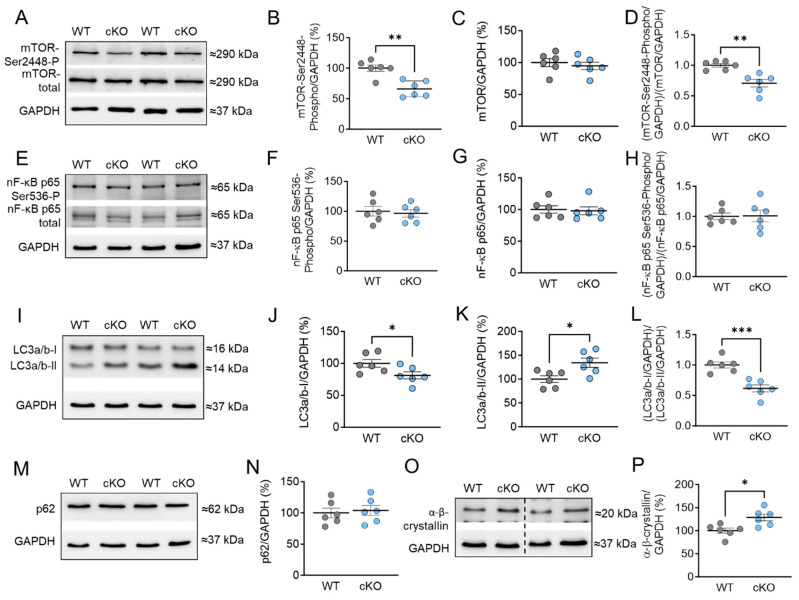
Protein kinase D (PKD) enzymes play important roles in regulating myocardial contraction, hypertrophy, and remodeling. One of the proteins phosphorylated by PKD is titin, which is involved in myofilament function. In this study, we aimed to investigate the role of PKD in cardiomyocyte function under conditions of oxidative stress. To do this, we used mice with a cardiomyocyte-specific knock-out of Prkd1, which encodes PKD1 (Prkd1loxP/loxP; αMHC-Cre; PKD1 cKO), as well as wild type littermate controls (Prkd1loxP/loxP; WT). We isolated permeabilized cardiomyocytes from PKD1 cKO mice and found that they exhibited increased passive stiffness (Fpassive), which was associated with increased oxidation of titin, but showed no change in titin ubiquitination. Additionally, the PKD1 cKO mice showed increased myofilament calcium (Ca2+) sensitivity (pCa50) and reduced maximum Ca2+-activated tension. These changes were accompanied by increased oxidation and reduced phosphorylation of the small myofilament protein cardiac myosin binding protein C (cMyBPC), as well as altered phosphorylation levels at different phosphosites in troponin I (TnI). The increased Fpassive and pCa50, and the reduced maximum Ca2+-activated tension were reversed when we treated the isolated permeabilized cardiomyocytes with reduced glutathione (GSH). This indicated that myofilament protein oxidation contributes to cardiomyocyte dysfunction. Furthermore, the PKD1 cKO mice exhibited increased oxidative stress and increased expression of pro-inflammatory markers interleukin (IL)-6, IL-18, and tumor necrosis factor alpha (TNF-α). Both oxidative stress and inflammation contributed to an increase in microtubule-associated protein 1 light chain 3 (LC3)-II levels and heat shock response by inhibiting the mammalian target of rapamycin (mTOR) in the PKD1 cKO mouse myocytes. These findings revealed a previously unknown role for PKD1 in regulating diastolic passive properties, myofilament Ca2+ sensitivity, and maximum Ca2+-activated tension under conditions of oxidative stress. Finally, we emphasized the importance of PKD1 in maintaining the balance of oxidative stress and inflammation in the context of autophagy, as well as cardiomyocyte function.
Keywords: autophagy; inflammation; myofilament proteins; protein kinase D; protein oxidation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Modulation of Titin-Based Stiffness in Hypertrophic Cardiomyopathy via Protein Kinase D.Front Physiol. 2020 Apr 15;11:240. doi: 10.3389/fphys.2020.00240. eCollection 2020. Front Physiol. 2020. PMID: 32351396 Free PMC article.
-
Diabetes-Induced Cardiomyocyte Passive Stiffening Is Caused by Impaired Insulin-Dependent Titin Modification and Can Be Modulated by Neuregulin-1.Circ Res. 2018 Jul 20;123(3):342-355. doi: 10.1161/CIRCRESAHA.117.312166. Epub 2018 May 14. Circ Res. 2018. PMID: 29760016
-
Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction.Cardiovasc Res. 2013 Mar 1;97(3):464-71. doi: 10.1093/cvr/cvs353. Epub 2012 Dec 4. Cardiovasc Res. 2013. PMID: 23213108
-
Emerging importance of oxidative stress in regulating striated muscle elasticity.J Muscle Res Cell Motil. 2015 Feb;36(1):25-36. doi: 10.1007/s10974-014-9392-y. Epub 2014 Nov 6. J Muscle Res Cell Motil. 2015. PMID: 25373878 Free PMC article. Review.
-
Myofilament dysfunction in cardiac disease from mice to men.J Muscle Res Cell Motil. 2008;29(6-8):189-201. doi: 10.1007/s10974-008-9160-y. Epub 2009 Jan 13. J Muscle Res Cell Motil. 2008. PMID: 19140019 Review.
Cited by
-
Diabetes mellitus aggravates myocardial inflammation and oxidative stress in aortic stenosis: a mechanistic link to HFpEF features.Cardiovasc Diabetol. 2025 May 13;24(1):203. doi: 10.1186/s12933-025-02748-y. Cardiovasc Diabetol. 2025. PMID: 40361188 Free PMC article.
-
Dysregulated inflammation, oxidative stress, and protein quality control in diabetic HFpEF: unraveling mechanisms and therapeutic targets.Cardiovasc Diabetol. 2025 May 14;24(1):211. doi: 10.1186/s12933-025-02734-4. Cardiovasc Diabetol. 2025. PMID: 40369521 Free PMC article.
References
-
- Dirkx E., Schwenk R.W., Coumans W.A., Hoebers N., Angin Y., Viollet B., Bonen A., Van Eys G.J.J.M., Glatz J.F.C., Luiken J.J.F.P. Protein Kinase D1 Is Essential for Contraction-Induced Glucose Uptake but Is Not Involved in Fatty Acid Uptake into Cardiomyocytes. J. Biol. Chem. 2012;287:5871–5881. doi: 10.1074/jbc.M111.281881. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
