

Mowat-Wilson Syndrome: Case Report and Review of ZEB2 Gene Variant Types, Protein Defects and Molecular Interactions
- PMID: 38474085
- PMCID: PMC10932183
- DOI: 10.3390/ijms25052838
Mowat-Wilson Syndrome: Case Report and Review of ZEB2 Gene Variant Types, Protein Defects and Molecular Interactions
Abstract
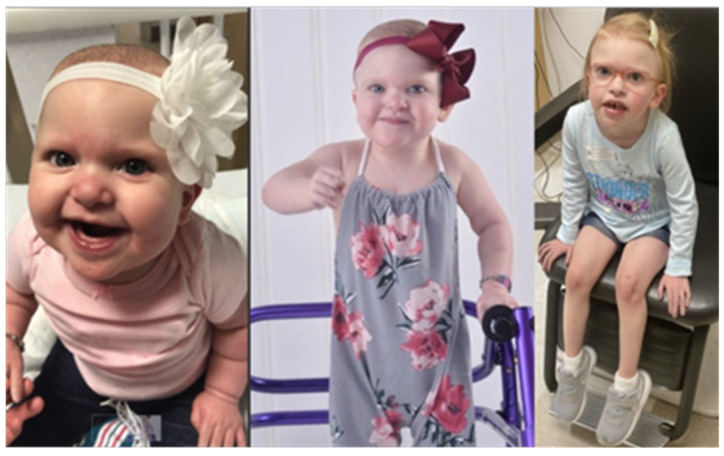
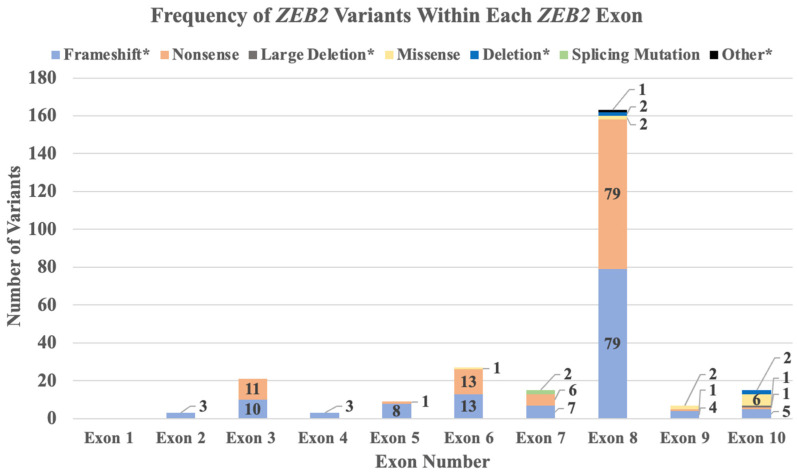
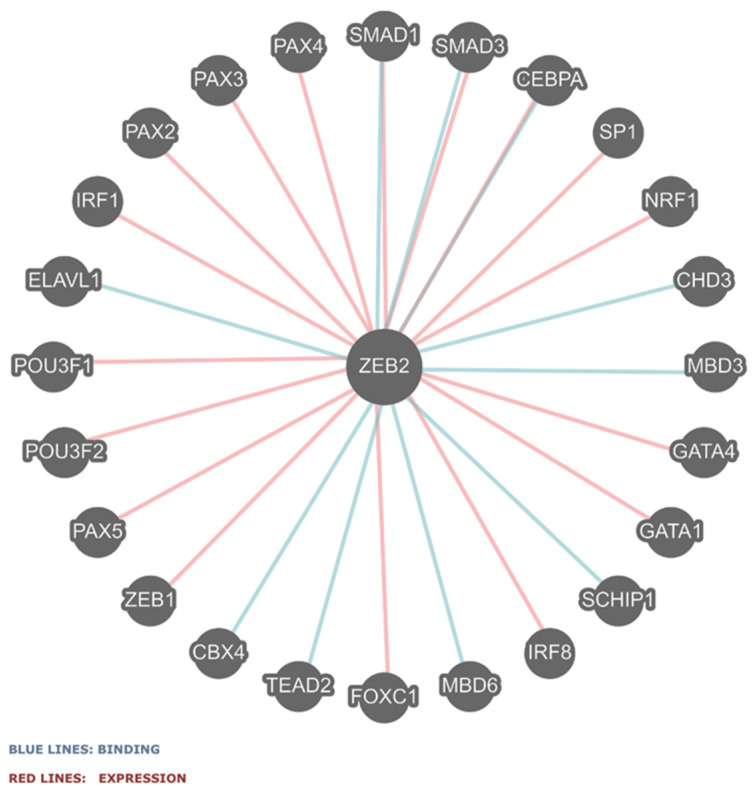
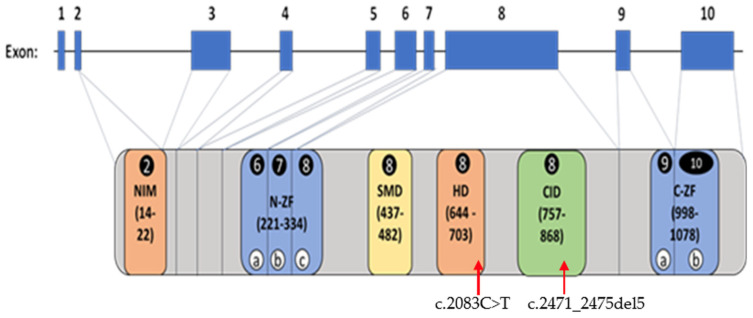
Mowat-Wilson syndrome (MWS) is a rare genetic neurodevelopmental congenital disorder associated with various defects of the zinc finger E-box binding homeobox 2 (ZEB2) gene. The ZEB2 gene is autosomal dominant and encodes six protein domains including the SMAD-binding protein, which functions as a transcriptional corepressor involved in the conversion of neuroepithelial cells in early brain development and as a mediator of trophoblast differentiation. This review summarizes reported ZEB2 gene variants, their types, and frequencies among the 10 exons of ZEB2. Additionally, we summarized their corresponding encoded protein defects including the most common variant, c.2083 C>T in exon 8, which directly impacts the homeodomain (HD) protein domain. This single defect was found in 11% of the 298 reported patients with MWS. This review demonstrates that exon 8 encodes at least three of the six protein domains and accounts for 66% (198/298) of the variants identified. More than 90% of the defects were due to nonsense or frameshift changes. We show examples of protein modeling changes that occurred as a result of ZEB2 gene defects. We also report a novel pathogenic variant in exon 8 in a 5-year-old female proband with MWS. This review further explores other genes predicted to be interacting with the ZEB2 gene and their predicted gene-gene molecular interactions with protein binding effects on embryonic multi-system development such as craniofacial, spine, brain, kidney, cardiovascular, and hematopoiesis.
Keywords: Mowat–Wilson syndrome (MWS); ZEB2 functional molecular interactions; ZEB2 gene variants; ZEB2 protein domains and defects; case report; review.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

References
-
- Mowat D.R., Croaker G.D., Cass D.T., Kerr B.A., Chaitow J., Adès L.C., Chia N.L., Wilson M.J. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: Delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J. Med. Genet. 1998;8:617–623. doi: 10.1136/jmg.35.8.617. - DOI - PMC - PubMed
-
- Cacheux V., Dastot-Le Moal F., Kääriäinen H., Bondurand N., Rintala R., Boissier B., Wilson M., Mowat D., Goossens M. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum. Mol. Genet. 2001;14:1503–1510. doi: 10.1093/hmg/10.14.1503. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
