

Semaglutide Improves Liver Steatosis and De Novo Lipogenesis Markers in Obese and Type-2-Diabetic Mice with Metabolic-Dysfunction-Associated Steatotic Liver Disease
- PMID: 38474208
- PMCID: PMC10932050
- DOI: 10.3390/ijms25052961
Semaglutide Improves Liver Steatosis and De Novo Lipogenesis Markers in Obese and Type-2-Diabetic Mice with Metabolic-Dysfunction-Associated Steatotic Liver Disease
Abstract
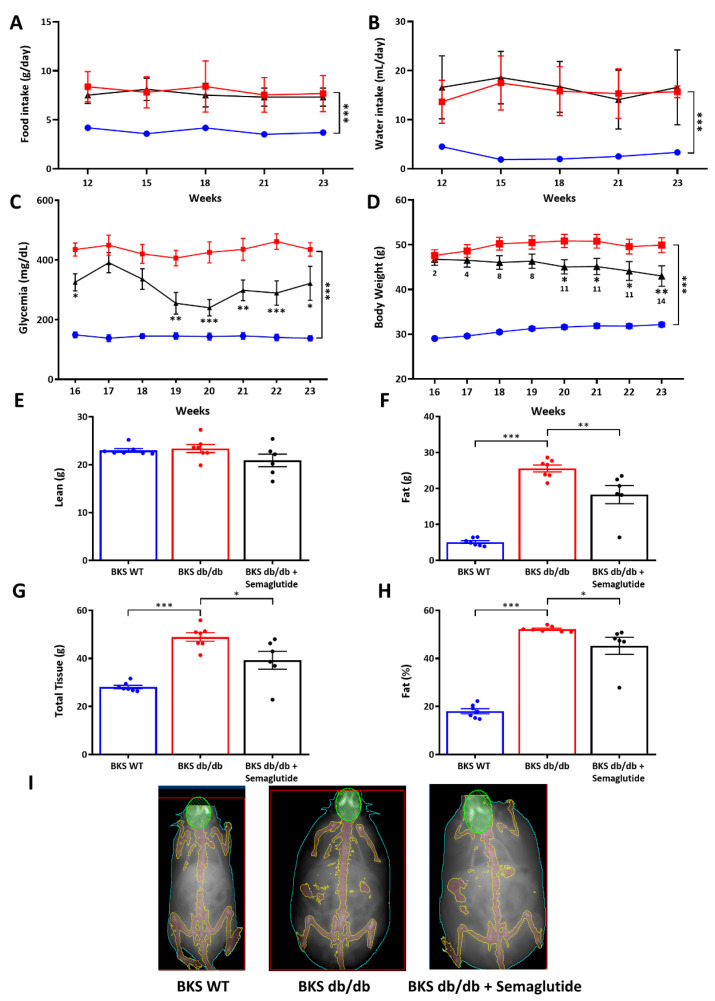
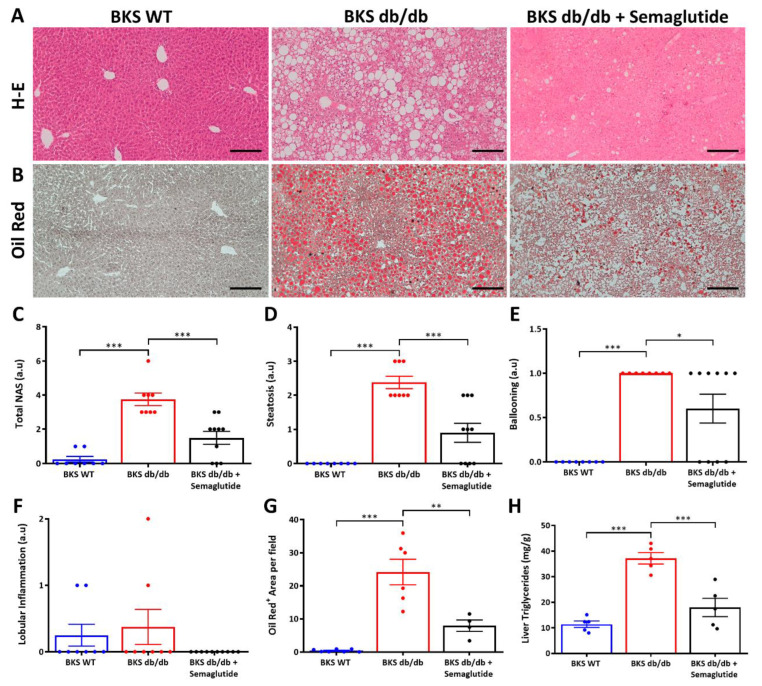
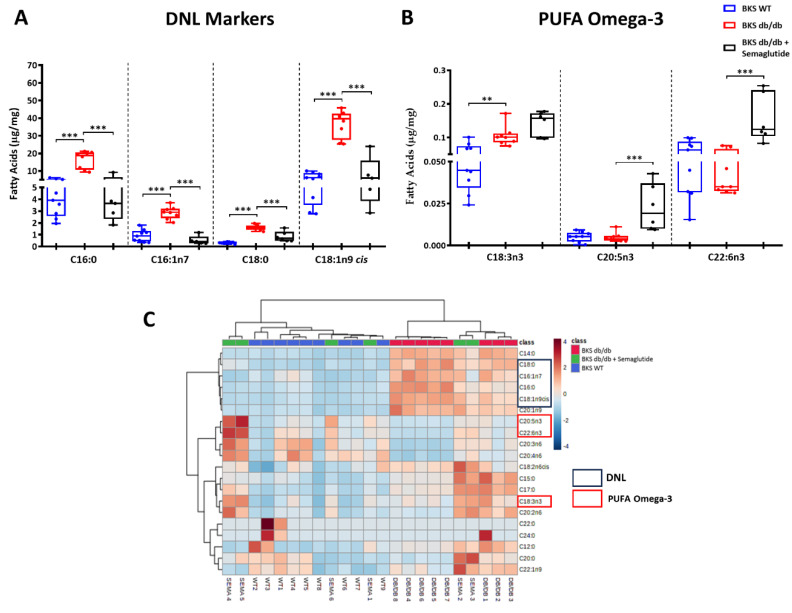
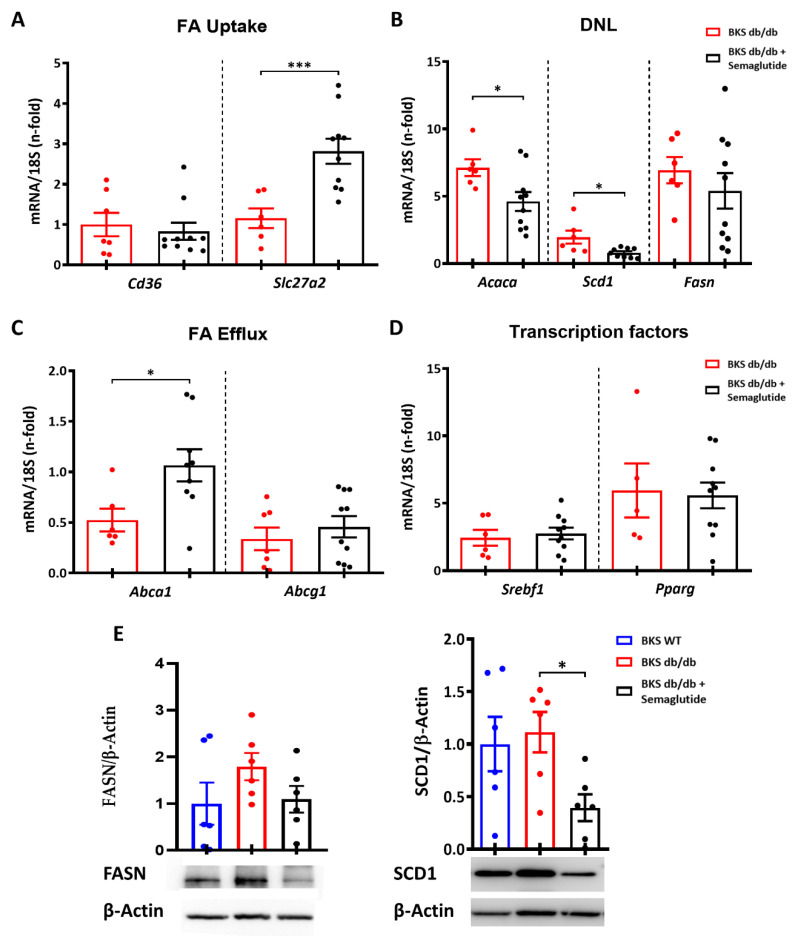
Metabolic-dysfunction-associated steatotic liver disease (MASLD) is a prevalent clinical condition associated with elevated morbidity and mortality rates. Patients with MASLD treated with semaglutide, a glucagon-like peptide-1 receptor agonist, demonstrate improvement in terms of liver damage. However, the mechanisms underlaying this beneficial effect are not yet fully elucidated. We investigated the efficacy of semaglutide in halting MASLD progression using a genetic mouse model of diabesity. Leptin-receptor-deficient mice with obesity and diabetes (BKS db/db) were either untreated or administered with semaglutide for 11 weeks. Changes in food and water intake, body weight and glycemia were monitored throughout the study. Body fat composition was assessed by dual-energy X-ray absorptiometry. Upon sacrifice, serum biochemical parameters, liver morphology, lipidomic profile and liver-lipid-related pathways were evaluated. The semaglutide-treated mice exhibited lower levels of glycemia, body weight, serum markers of liver dysfunction and total and percentage of fat mass compared to untreated db/db mice without a significant reduction in food intake. Histologically, semaglutide reduced hepatic steatosis, hepatocellular ballooning and intrahepatic triglycerides. Furthermore, the treatment ameliorated the hepatic expression of de novo lipogenesis markers and modified lipid composition by increasing the amount of polyunsaturated fatty acids. The administration of semaglutide to leptin-receptor-deficient, hyperphagic and diabetic mice resulted in the amelioration of MASLD, likely independently of daily caloric intake, suggesting a direct effect of semaglutide on the liver through modulation of the lipid profile.
Keywords: GLP1 receptor agonists; diabetes; insulin resistance; obesity; semaglutide; steatosis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Effects of NPY-2 Receptor Antagonists, Semaglutide, PYY3-36, and Empagliflozin on Early MASLD in Diet-Induced Obese Rats.Nutrients. 2024 Mar 21;16(6):904. doi: 10.3390/nu16060904. Nutrients. 2024. PMID: 38542814 Free PMC article.
-
Chronic treatment with glucagon-like peptide-1 and glucagon receptor co-agonist causes weight loss-independent improvements in hepatic steatosis in mice with diet-induced obesity.Biomed Pharmacother. 2024 Jul;176:116888. doi: 10.1016/j.biopha.2024.116888. Epub 2024 Jun 10. Biomed Pharmacother. 2024. PMID: 38861859
-
Evaluation of long acting GLP1R/GCGR agonist in a DIO and biopsy-confirmed mouse model of NASH suggest a beneficial role of GLP-1/glucagon agonism in NASH patients.Mol Metab. 2024 Jan;79:101850. doi: 10.1016/j.molmet.2023.101850. Epub 2023 Dec 7. Mol Metab. 2024. PMID: 38065435 Free PMC article.
-
Influence of Lipid Class Used for Omega-3 Fatty Acid Supplementation on Liver Fat Accumulation in MASLD.Physiol Res. 2024 Aug 31;73(Suppl 1):S295-S320. doi: 10.33549/physiolres.935396. Epub 2024 Jul 17. Physiol Res. 2024. PMID: 39016154 Free PMC article. Review.
-
Hepatic fatty acid and glucose handling in metabolic disease: Potential impact on cardiovascular disease risk.Atherosclerosis. 2024 Jul;394:117237. doi: 10.1016/j.atherosclerosis.2023.117237. Epub 2023 Aug 11. Atherosclerosis. 2024. PMID: 37633797 Review.
Cited by
-
Exploring the Protective Effects of Traditional Antidiabetic Medications and Novel Antihyperglycemic Agents in Diabetic Rodent Models.Pharmaceuticals (Basel). 2025 May 1;18(5):670. doi: 10.3390/ph18050670. Pharmaceuticals (Basel). 2025. PMID: 40430489 Free PMC article. Review.
-
Development of SOCS1 mimetics as novel approach to harmonize inflammation, oxidative stress, and fibrogenesis in metabolic dysfunction-associated steatotic liver disease.Redox Biol. 2025 Jul;84:103670. doi: 10.1016/j.redox.2025.103670. Epub 2025 May 11. Redox Biol. 2025. PMID: 40373621 Free PMC article.
-
Association of Neutrophil/High-Density Lipoprotein Cholesterol Ratio with Metabolic Dysfunction-Associated Steatotic Liver Disease in Type 2 Diabetes Mellitus Patients: A Cross-Sectional Study and Predictive Model Construction.Diabetes Metab Syndr Obes. 2025 Jul 29;18:2597-2609. doi: 10.2147/DMSO.S528506. eCollection 2025. Diabetes Metab Syndr Obes. 2025. PMID: 40755748 Free PMC article.
-
Semaglutide Treatment Effects on Liver Fat Content in Obese Subjects with Metabolic-Associated Steatotic Liver Disease (MASLD).J Clin Med. 2024 Oct 13;13(20):6100. doi: 10.3390/jcm13206100. J Clin Med. 2024. PMID: 39458050 Free PMC article.
-
Semaglutide as a GLP-1 Agonist: A Breakthrough in Obesity Treatment.Pharmaceuticals (Basel). 2025 Mar 12;18(3):399. doi: 10.3390/ph18030399. Pharmaceuticals (Basel). 2025. PMID: 40143174 Free PMC article. Review.
References
-
- Riazi K., Azhari H., Charette J.H., Underwood F.E., King J.A., Afshar E.E., Swain M.G., Congly S.E., Kaplan G.G., Shaheen A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022;7:851–861. doi: 10.1016/S2468-1253(22)00165-0. - DOI - PubMed
-
- Rinella M.E., Lazarus J.V., Ratziu V., Francque S.M., Sanyal A.J., Kanwal F., Romero D., Abdelmalek M.F., Anstee Q.M., Arab J.P., et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;29:101133.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
