

Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study
- PMID: 38474554
- PMCID: PMC10933821
- DOI: 10.3390/molecules29051043
Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study
Abstract
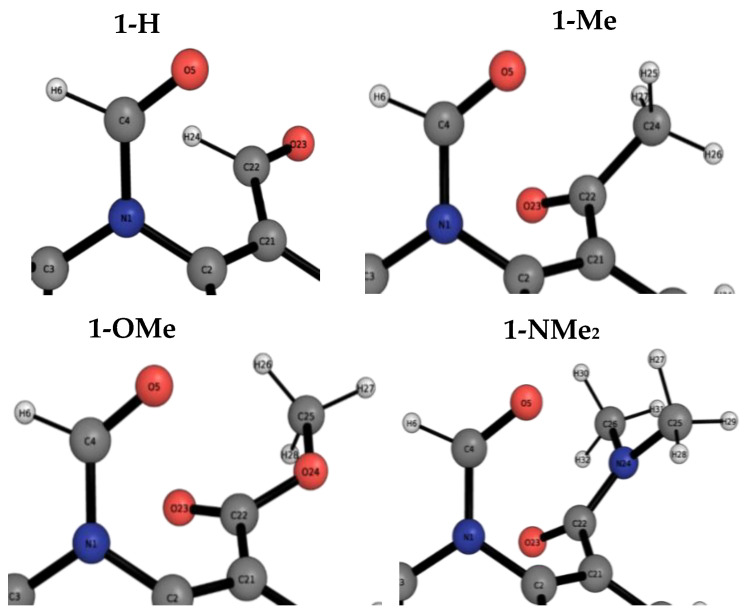
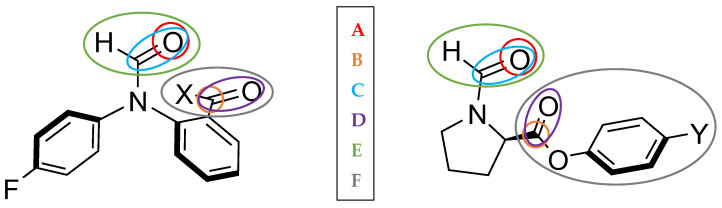
The interaction energies of two series of molecular balances (1-X with X = H, Me, OMe, NMe2 and 2-Y with Y = H, CN, NO2, OMe, NMe2) designed to probe carbonyl…carbonyl interactions were analysed at the B3LYP/6-311++G(d,p)-D3 level of theory using the energy partitioning method of Interacting Quantum Atoms/Fragments (IQA/IQF). The partitioned energies are analysed by the Relative Energy Gradient (REG) method, which calculates the correlation between these energies and the total energy of a system, thereby explaining the role atoms have in the energetic behaviour of the total system. The traditional "back-of-the-envelope" open and closed conformations of molecular balances do not correspond to those of the lowest energy. Hence, more care needs to be taken when considering which geometries to use for comparison with the experiment. The REG-IQA method shows that the 1-H and 1-OMe balances behave differently to the 1-Me and 1-NMe2 balances because the latter show more prominent electrostatics between carbonyl groups and undergoes a larger dihedral rotation due to the bulkiness of the functional groups. For the 2-Y balance, REG-IQA shows the same behaviour across the series as the 1-H and 1-OMe balances. From an atomistic point of view, the formation of the closed conformer is favoured by polarisation and charge-transfer effects on the amide bond across all balances and is counterbalanced by a de-pyramidalisation of the amide nitrogen. Moreover, focusing on the oxygen of the amide carbonyl and the α-carbon of the remaining carbonyl group, electrostatics have a major role in the formation of the closed conformer, which goes against the well-known n-π* interaction orbital overlap concept. However, REG-IQF shows that exchange-correlation energies overtake electrostatics for all the 2-Y balances when working with fragments around the carbonyl groups, while they act on par with electrostatics for the 1-OMe and 1-NMe2. REG-IQF also shows that exchange-correlation energies in the 2-Y balance are correlated to the inductive electron-donating and -withdrawing trends on aromatic groups. We demonstrate that methods such as REG-IQA/IQF can help with the fine-tuning of molecular balances prior to the experiment and that the energies that govern the probed interactions are highly dependent on the atoms and functional groups involved.
Keywords: DFT; IQA; QTAIM; Relative Energy Gradient (REG); conformational analysis; molecular balances; quantum chemical topology (QCT).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures





Similar articles
-
Directionality of Halogen Bonds: An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study.Chemphyschem. 2019 Aug 5;20(15):1922-1930. doi: 10.1002/cphc.201900250. Epub 2019 Jul 5. Chemphyschem. 2019. PMID: 31136067
-
An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation.Molecules. 2020 Jun 9;25(11):2674. doi: 10.3390/molecules25112674. Molecules. 2020. PMID: 32526931 Free PMC article.
-
The IQA Energy Partition in a Drug Design Setting: A Hepatitis C Virus RNA-Dependent RNA Polymerase (NS5B) Case Study.Pharmaceuticals (Basel). 2022 Oct 8;15(10):1237. doi: 10.3390/ph15101237. Pharmaceuticals (Basel). 2022. PMID: 36297349 Free PMC article.
-
A combined BET and IQA-REG study of the activation energy of non-polar zw-type [3+2] cycloaddition reactions.Phys Chem Chem Phys. 2023 Apr 12;25(15):10853-10865. doi: 10.1039/d3cp00329a. Phys Chem Chem Phys. 2023. PMID: 37013716
-
Interacting Quantum Atoms-A Review.Molecules. 2020 Sep 3;25(17):4028. doi: 10.3390/molecules25174028. Molecules. 2020. PMID: 32899346 Free PMC article. Review.
References
-
- Kollman P.A. Noncovalent interactions. Acc. Chem. Res. 1977;10:365–371. doi: 10.1021/ar50118a003. - DOI
-
- London F. The general theory of molecular forces. Trans. Faraday Soc. 1937;33:8b–26. doi: 10.1039/tf937330008b. - DOI
-
- Dzyaloshinskii I.E., Lifshitz E.M., Pitaevskii L.P. The general theory of van der Waals forces. Adv. Phys. 1961;10:165–209. doi: 10.1080/00018736100101281. - DOI
-
- Hobza P., Müller-Dethlefs K. Non-Covalent Interactions: Theory and Experiment. Volume 2 Royal Society of Chemistry; London, UK: 2010.
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
