

MetaQuad: shared informative variants discovery in metagenomic samples
- PMID: 38476299
- PMCID: PMC10932609
- DOI: 10.1093/bioadv/vbae030
MetaQuad: shared informative variants discovery in metagenomic samples
Abstract
Motivation: Strain-level analysis of metagenomic data has garnered significant interest in recent years. Microbial single nucleotide polymorphisms (SNPs) are genomic variants that can reflect strain-level differences within a microbial species. The diversity and emergence of SNPs in microbial genomes may reveal evolutionary history and environmental adaptation in microbial populations. However, efficient discovery of shared polymorphic variants in a large collection metagenomic samples remains a computational challenge.
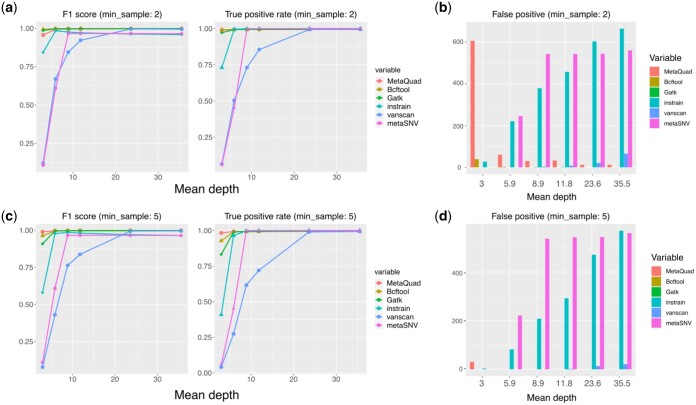
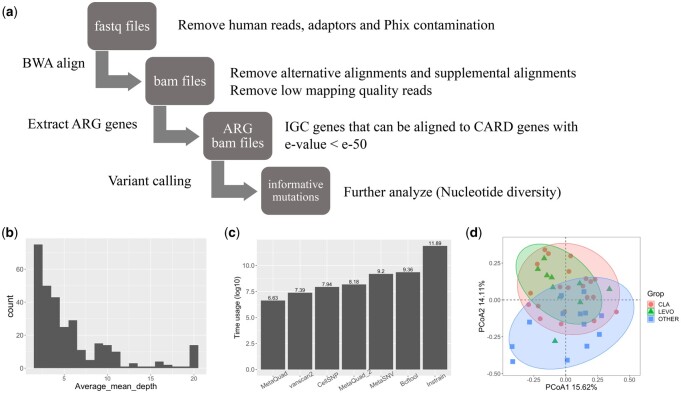
Results: MetaQuad utilizes a density-based clustering technique to effectively distinguish between shared variants and non-polymorphic sites using shotgun metagenomic data. Empirical comparisons with other state-of-the-art methods show that MetaQuad significantly reduces the number of false positive SNPs without greatly affecting the true positive rate. We used MetaQuad to identify antibiotic-associated variants in patients who underwent Helicobacter pylori eradication therapy. MetaQuad detected 7591 variants across 529 antibiotic resistance genes. The nucleotide diversity of some genes is increased 6 weeks after antibiotic treatment, potentially indicating the role of these genes in specific antibiotic treatments.
Availability and implementation: MetaQuad is an open-source Python package available via https://github.com/holab-hku/MetaQuad.
© The Author(s) 2024. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures

References
-
- Ankerst M, Breunig MM, Kriegel H-P. et al. OPTICS: ordering points to identify the clustering structure. SIGMOD Rec 1999;28:49–60.
-
- Bushnell B. BBMap: A Fast, Accurate, Splice-Aware Aligner. 2014. https://www.osti.gov/servlets/purl/1241166
LinkOut - more resources
Full Text Sources
