

The archaeome in metaorganism research, with a focus on marine models and their bacteria-archaea interactions
- PMID: 38476944
- PMCID: PMC10927989
- DOI: 10.3389/fmicb.2024.1347422
The archaeome in metaorganism research, with a focus on marine models and their bacteria-archaea interactions
Abstract
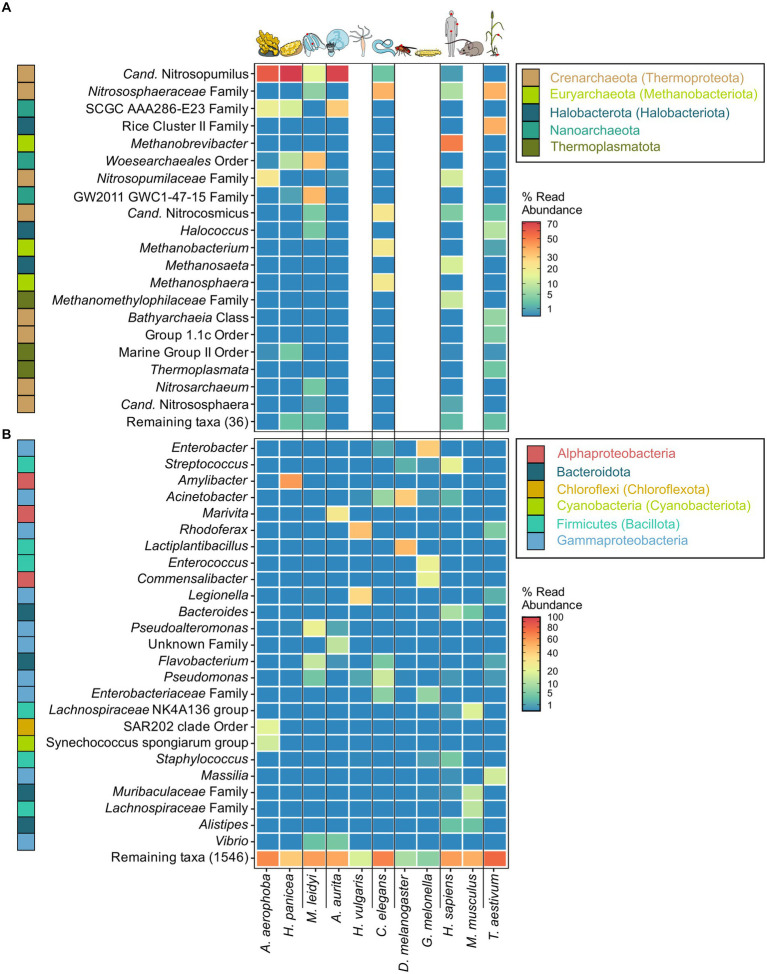
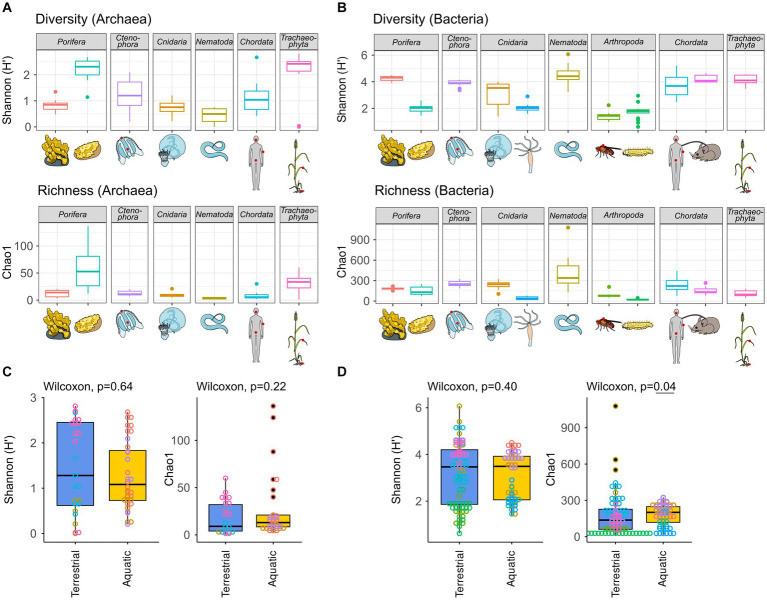
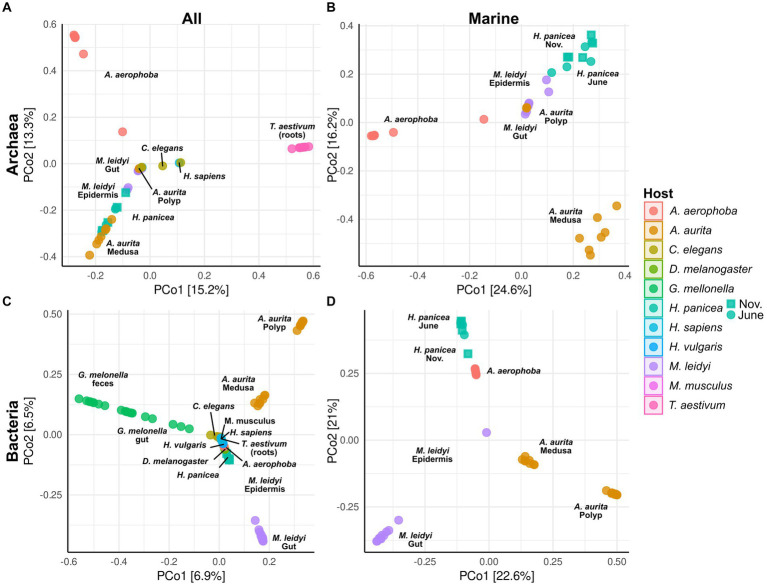
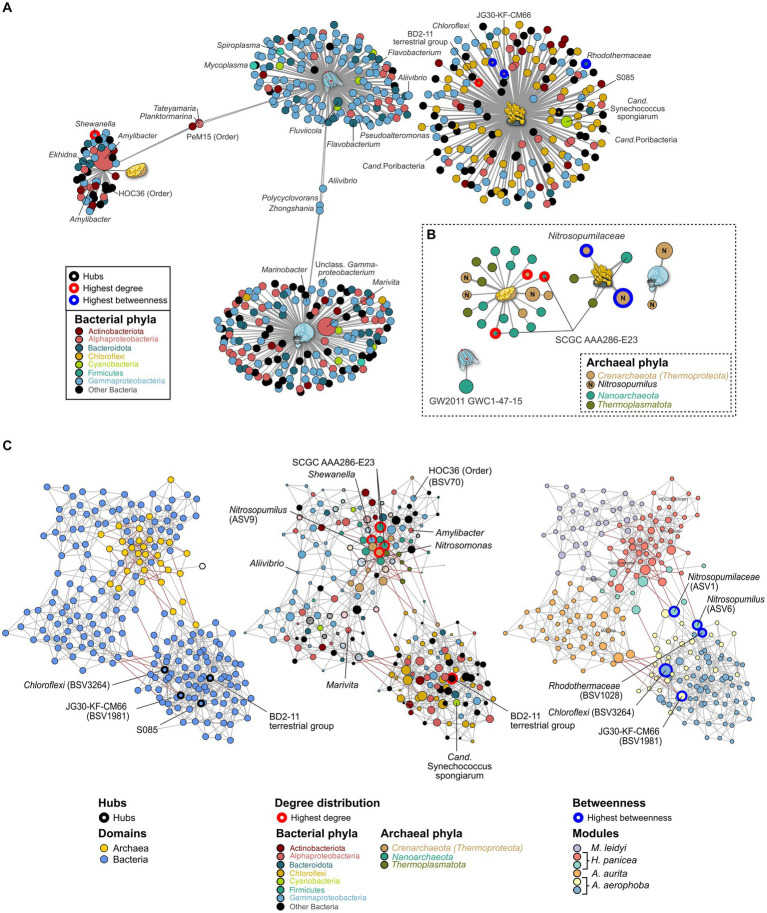
Metaorganism research contributes substantially to our understanding of the interaction between microbes and their hosts, as well as their co-evolution. Most research is currently focused on the bacterial community, while archaea often remain at the sidelines of metaorganism-related research. Here, we describe the archaeome of a total of eleven classical and emerging multicellular model organisms across the phylogenetic tree of life. To determine the microbial community composition of each host, we utilized a combination of archaea and bacteria-specific 16S rRNA gene amplicons. Members of the two prokaryotic domains were described regarding their community composition, diversity, and richness in each multicellular host. Moreover, association with specific hosts and possible interaction partners between the bacterial and archaeal communities were determined for the marine models. Our data show that the archaeome in marine hosts predominantly consists of Nitrosopumilaceae and Nanoarchaeota, which represent keystone taxa among the porifera. The presence of an archaeome in the terrestrial hosts varies substantially. With respect to abundant archaeal taxa, they harbor a higher proportion of methanoarchaea over the aquatic environment. We find that the archaeal community is much less diverse than its bacterial counterpart. Archaeal amplicon sequence variants are usually host-specific, suggesting adaptation through co-evolution with the host. While bacterial richness was higher in the aquatic than the terrestrial hosts, a significant difference in diversity and richness between these groups could not be observed in the archaeal dataset. Our data show a large proportion of unclassifiable archaeal taxa, highlighting the need for improved cultivation efforts and expanded databases.
Keywords: archaeome; host-associated microbiota; marine archaea; metaorganism; microbial community; microbiome.
Copyright © 2024 von Hoyningen-Huene, Bang, Rausch, Rühlemann, Fokt, He, Jensen, Knop, Petersen, Schmittmann, Zimmer, Baines, Bosch, Hentschel, Reusch, Roeder, Franke, Schulenburg, Stukenbrock and Schmitz.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Andersen K. S., Kirkegaard R. H., Karst S. M., Albertsen M. (2018). ampvis2: an R package to analyse and visualise 16S rRNA amplicon data. bioRxiv:299537. doi: 10.1101/299537 - DOI
-
- Barnett D. J. M., Arts I. C. W., Penders J. (2021). microViz: an R package for microbiome data visualization and statistics. J. Open Source Softw. 6:3201. doi: 10.21105/joss.03201 - DOI
LinkOut - more resources
Full Text Sources