

Contributions of Common Genetic Variants to Constitutional Delay of Puberty and Idiopathic Hypogonadotropic Hypogonadism
- PMID: 38477512
- PMCID: PMC11651688
- DOI: 10.1210/clinem/dgae166
Contributions of Common Genetic Variants to Constitutional Delay of Puberty and Idiopathic Hypogonadotropic Hypogonadism
Abstract
Context: Constitutional delay of puberty (CDP) is highly heritable, but the genetic basis for CDP is largely unknown. Idiopathic hypogonadotropic hypogonadism (IHH) can be caused by rare genetic variants, but in about half of cases, no rare-variant cause is found.
Objective: To determine whether common genetic variants that influence pubertal timing contribute to CDP and IHH.
Design: Case-control study.
Participants: 80 individuals with CDP; 301 with normosmic IHH, and 348 with Kallmann syndrome (KS); control genotyping data from unrelated studies.
Main outcome measures: Polygenic scores (PGS) based on genome-wide association studies for timing of male pubertal hallmarks and age at menarche (AAM).
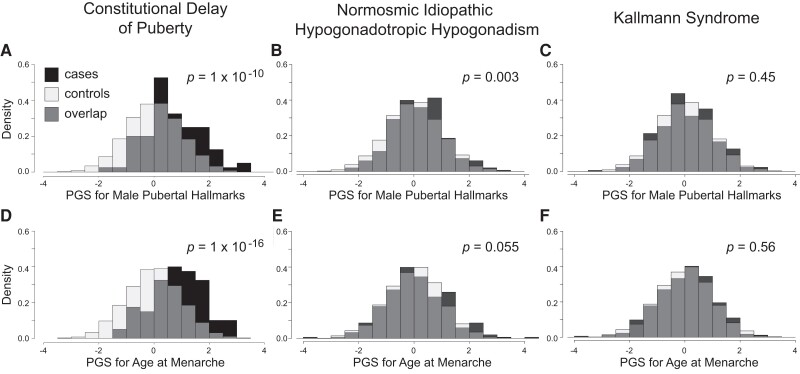
Results: The CDP cohort had higher PGS for male pubertal hallmarks and for AAM compared to controls (for male hallmarks, Cohen's d = 0.67, P = 1 × 10-10; for AAM, d = 0.85, P = 1 × 10-16). The normosmic IHH cohort also had higher PGS for male hallmarks compared to controls, but the difference was smaller (male hallmarks d = 0.20, P = .003; AAM d = 0.10, P = .055). No differences were seen for the KS cohort compared to controls (male hallmarks d = 0.05, P = .45; AAM d = 0.03, P = .56).
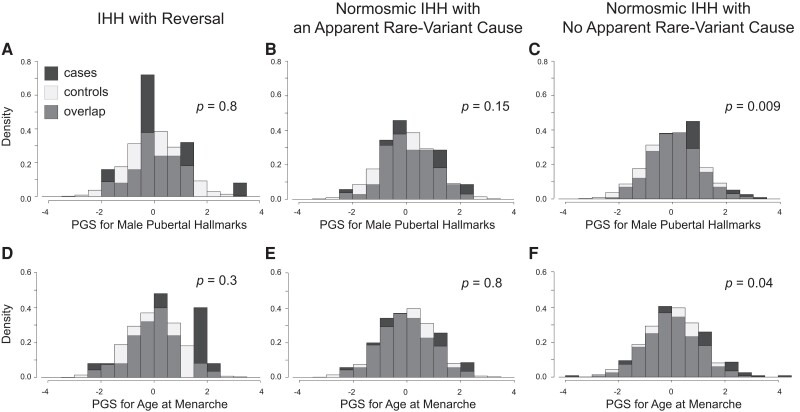
Conclusion: Common genetic variants that influence pubertal timing in the general population contribute strongly to the genetics of CDP, weakly to normosmic IHH, and potentially not at all to KS. These findings demonstrate that the common-variant genetics of CDP and normosmic IHH are largely but not entirely distinct.
Keywords: Kallmann syndrome; common genetic variants; constitutional delay of puberty; idiopathic hypogonadotropic hypogonadism; polygenic scores.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For commercial re-use, please contact reprints@oup.com for reprints and translation rights for reprints. All other permissions can be obtained through our RightsLink service via the Permissions link on the article page on our site—for further information please contact journals.permissions@oup.com.
Figures
References
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
