

Bub1 suppresses inflammatory arthritis-associated bone loss in mice through inhibition of TNFα-mediated osteoclastogenesis
- PMID: 38477771
- PMCID: PMC11240161
- DOI: 10.1093/jbmr/zjae015
Bub1 suppresses inflammatory arthritis-associated bone loss in mice through inhibition of TNFα-mediated osteoclastogenesis
Abstract
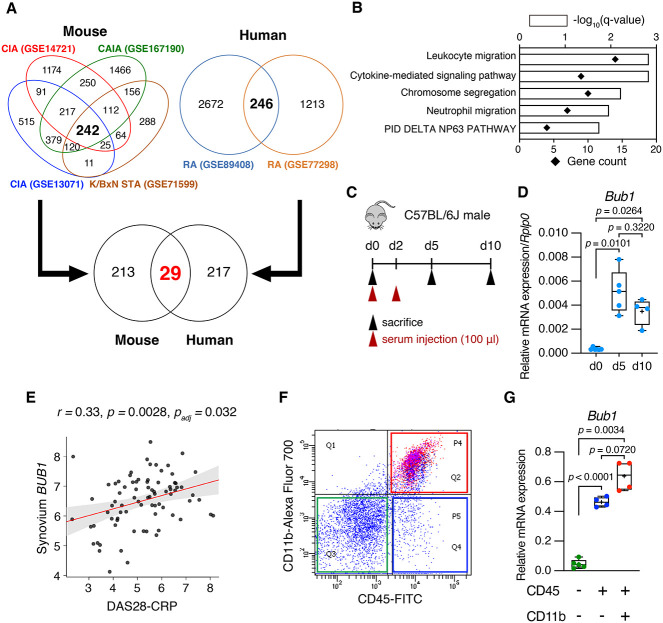
Rheumatoid arthritis (RA) is an inflammatory autoimmune disease characterized by synovitis, bone and cartilage destruction, and increased fracture risk with bone loss. Although disease-modifying antirheumatic drugs have dramatically improved clinical outcomes, these therapies are not universally effective in all patients because of the heterogeneity of RA pathogenesis. Therefore, it is necessary to elucidate the molecular mechanisms underlying RA pathogenesis, including associated bone loss, in order to identify novel therapeutic targets. In this study, we found that Budding uninhibited by benzimidazoles 1 (BUB1) was highly expressed in RA patients' synovium and murine ankle tissue with arthritis. As CD45+CD11b+ myeloid cells are a Bub1 highly expressing population among synovial cells in mice, myeloid cell-specific Bub1 conditional knockout (Bub1ΔLysM) mice were generated. Bub1ΔLysM mice exhibited reduced femoral bone mineral density when compared with control (Ctrl) mice under K/BxN serum-transfer arthritis, with no significant differences in joint inflammation or bone erosion based on a semi-quantitative erosion score and histological analysis. Bone histomorphometry revealed that femoral bone mass of Bub1ΔLysM under arthritis was reduced by increased osteoclastic bone resorption. RNA-seq and subsequent Gene Set Enrichment Analysis demonstrated a significantly enriched nuclear factor-kappa B pathway among upregulated genes in receptor activator of nuclear factor kappa B ligand (RANKL)-stimulated bone marrow-derived macrophages (BMMs) obtained from Bub1ΔLysM mice. Indeed, osteoclastogenesis using BMMs derived from Bub1ΔLysM was enhanced by RANKL and tumor necrosis factor-α or RANKL and IL-1β treatment compared with Ctrl. Finally, osteoclastogenesis was increased by Bub1 inhibitor BAY1816032 treatment in BMMs derived from wildtype mice. These data suggest that Bub1 expressed in macrophages plays a protective role against inflammatory arthritis-associated bone loss through inhibition of inflammation-mediated osteoclastogenesis.
Keywords: Bub1; NF-κB; bone metabolism; osteoclasts; rheumatoid arthritis.
Plain language summary
Rheumatoid arthritis (RA) is a disease caused by an abnormal immune system, resulting in inflammation, swelling, and bone destruction in the joints, along with systemic bone loss. While new medications have dramatically improved treatment efficacy, these therapies are not universally effective for all patients. Therefore, we need to understand the regulatory mechanisms behind RA, including associated bone loss, to develop better therapies. In this study, we found that Budding uninhibited by benzimidazoles 1 (Bub1) was highly expressed in inflamed joints, especially in myeloid cells, which are a type of immune cells. To explore its role, we created myeloid cell–specific Bub1 conditional knockout (cKO) mice and induced arthritis to analyze its role during arthritis. The cKO mice exhibited lower bone mineral density when compared with control mice under inflammatory arthritis because of increased osteoclastic bone resorption, without significant differences in joint inflammation or bone erosion. Further investigation showed that Bub1 prevents excessive osteoclast differentiation induced by inflammation in bone marrow macrophages. These data suggest that Bub1 in macrophages protects against bone loss caused by inflammatory arthritis, offering potential insights for developing treatments that focus on bone health.
© The Author(s) 2024. Published by Oxford University Press on behalf of the American Society for Bone and Mineral Research.
Conflict of interest statement
None declared.
Figures






References
-
- McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;55(12):2255–2270. - PubMed
-
- Haugeberg G, Uhlig T, Falch JA, Halse JI, Kvien TK. Bone mineral density and frequency of osteoporosis in female patients with rheumatoid arthritis: results from 394 patients in the Oslo County rheumatoid arthritis register. Arthritis Rheum. 2000;43(3):522–530. 10.1002/1529-0131(200003)43:3<522::AID-ANR7>3.0.CO;2-Y. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
