

Evolutionary trajectories of small cell lung cancer under therapy
- PMID: 38480884
- PMCID: PMC10972747
- DOI: 10.1038/s41586-024-07177-7
Evolutionary trajectories of small cell lung cancer under therapy
Abstract
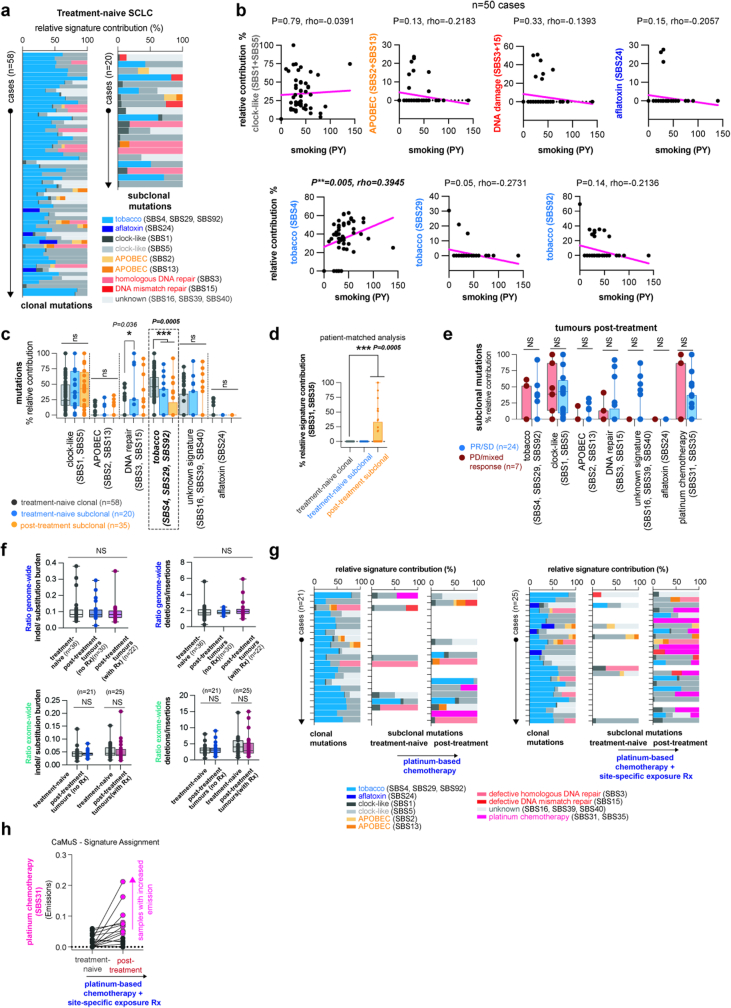
The evolutionary processes that underlie the marked sensitivity of small cell lung cancer (SCLC) to chemotherapy and rapid relapse are unknown1-3. Here we determined tumour phylogenies at diagnosis and throughout chemotherapy and immunotherapy by multiregion sequencing of 160 tumours from 65 patients. Treatment-naive SCLC exhibited clonal homogeneity at distinct tumour sites, whereas first-line platinum-based chemotherapy led to a burst in genomic intratumour heterogeneity and spatial clonal diversity. We observed branched evolution and a shift to ancestral clones underlying tumour relapse. Effective radio- or immunotherapy induced a re-expansion of founder clones with acquired genomic damage from first-line chemotherapy. Whereas TP53 and RB1 alterations were exclusively part of the common ancestor, MYC family amplifications were frequently not constituents of the founder clone. At relapse, emerging subclonal mutations affected key genes associated with SCLC biology, and tumours harbouring clonal CREBBP/EP300 alterations underwent genome duplications. Gene-damaging TP53 alterations and co-alterations of TP53 missense mutations with TP73, CREBBP/EP300 or FMN2 were significantly associated with shorter disease relapse following chemotherapy. In summary, we uncover key processes of the genomic evolution of SCLC under therapy, identify the common ancestor as the source of clonal diversity at relapse and show central genomic patterns associated with sensitivity and resistance to chemotherapy.
© 2024. The Author(s).
Conflict of interest statement
R.K.T. is a founder of, and consultant to, PearlRiver Bio, acquired by Centessa, and a shareholder of Centessa; a founder and shareholder of, and consultant to, Epiphanes; a founder, shareholder and CEO of DISCO Pharmaceuticals. M.P. is consultant to DISCO Pharmaceuticals. J.G. is consultant to DISCO Pharmaceuticals and received honoraria from MSD. J.W. participated in advisory boards and received lecture fees from Abion, Amgen, AstraZeneca, Bayer, Blueprint, BMS, Boehringer Ingelheim, Chugai, Daiichi Sankyo, Janssen, Lilly, Loxo, Merck, Mirati, MSD, Novartis, Nuvalent, Pfizer, Pierre-Fabre, Roche, Seattle Genetics, Takeda and Turning Point; and additional research support from BMS, Janssen Pharmaceutica, Novartis and Pfizer. M. Schuler is consultant to Amgen, AstraZeneca, Blueprint Medicines, Boehringer Ingelheim, Bristol Myers Squibb, GlaxoSmithKline, Janssen, Merck Serono, Novartis, Roche, Sanofi, Takeda, received honoraria from Amgen, Boehringer Ingelheim, Bristol Myers Squibb, Janssen, Novartis, Roche and Sanofi and received research funding from AstraZeneca and Bristol Myers Squibb. R.B. received honoraria for lectures and advisory boards for AbbVie, Amgen, AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Illumina, Janssen, Lilly, Merck Serono, MSD, Novartis, Qiagen, Pfizer, Roche and Targos MP/ Discovery Life Sciences. R.B. serves as a member of the board of directors and is a shareholder of Gnothis, Inc. (SE). A.Q. is consultant to Astellas, BMS, MSD, AstraZeneca, Servier and Oncowissen App/TV. R.N.F. received funding from BMS and MSD. J.K. received honoraria from Roche, Sanofi, Boehringer Ingelheim, MSD, BMS/Celgene, Lilly, Takeda, Novartis, AstraZeneca and Pfizer; is consultant to Roche, Boehringer Ingelheim, MSD, BMS/Celgene, Lilly, Takeda, Novartis, AstraZeneca, Pfizer and Janssen; and received travel and congress support from Roche, Sanofi, Boehringer Ingelheim, MSD, BMS/Celgene, Lilly, Takeda, Novartis, AstraZeneca, Pfizer and Janssen. H.-D.H. has participated in advisory board meetings of Amgen and Boehringer Ingelheim, is a steering committee member for Amgen and coordinating or local PI for Amgen, BMS, Revolution Medicines, Boehringer Ingelheim, Merck, Norvatis, AstraZeneca, Dracen, Daiichi Sankyo and AIO-Studien-gGmbH and reports personal fees from Amgen, BMS and Johnson & Johnson. J.P.P. reports personal fees from Apellis/Sobi, Alexion/AstraZeneca, Amgen, Blueprint Medicines, BMS, Boehringer Ingelheim, Gilead, MSD, Novartis, Pfizer, Roche, Sanofi and SwixxBiopharma outside the submitted work. M.K. is consultant to Boehringer Ingelheim, Bayer, BMS, Chugai and Roche and received honoraria from Novartis and Boehringer Ingelheim. L.H. received honoraria and personal fees from Boehringer Ingelheim, BMS, Pfizer, Roche and AstraZeneca. F.J. received scientific support from Merck and AstraZeneca. V.H. is a shareholder of BMS and Johnson & Johnson and received honoraria from Roche, Amgen, Pfizer, Celgene, BMS, Boehringer, Novartis, AstraZeneca and Lilly. F.G. received funding from ASTRA, Boehringer Ingelheim, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Siemens and GSK; has been appointed speaker for ASTRA, Boehringer, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Ariad, Abbvie, Siemens, Tesaro/GSK, Amgen and Daiichi Sankyo; and participates in administrative boards for ASTRA, Boehringer, BMS, Celgene, Lilly, MSD, Novartis, Pfizer, Roche, Takeda, Ariad, Abbvie, Tesaro/GSK, Siemens, Tesaro, Amgen and Daiichi Sankyo. C.G. received honoraria from Roche, Sanofi, Boehringer Ingelheim, MSD, BMS/Celgene, Lilly, Takeda, Novartis, AstraZeneca and Pfizer; is consultant to Roche, Boehringer Ingelheim, MSD, BMS/Celgene, Lilly, Takeda, Novartis, AstraZeneca, Pfizer and Janssen; and has received travel and congress support from Boehringer Ingelheim, MSD, Lilly, Takeda, Novartis, AstraZeneca, Pfizer and Janssen. R.T.U. received honoraria from Roche, Boehringer Ingelheim, GSK, PharmaMar and Bayer. C.A. participated on advisory boards and received honoraria from AstraZeneca, Biotest, Bristol Myers Squibb, MSD and Roche; and is a consultant to Ewimed and received research support from Bristol Myers Squibb and PharmaCep—all outside of the submitted work. M. Sebastian reports grants from AstraZeneca; consulting fees from AstraZeneca, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Lilly, Roche, Boehringer lngelheim, Amgen, Takeda, Johnson, Merck Serono and GSK; honoraria for lectures from AstraZeneca, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Lilly, Roche, Boehringer lngelheim, Amgen, Takeda, Johnson, CureVac, BioNTech, Merck Serono, GSK, Daiichi and Pfizer; travel support from Takeda and Pfizer; and membership of the advisory board of CureVac and BioNTech. J.A.S. reports personal fees from Boehringer Ingelheim, AstraZeneca, Roche, BMS, Amgen, LEO pharma, Novartis and Takeda—all outside of the submitted work. All other authors report no competing interests.
Figures













References
-
- Zugazagoitia, J. & Paz-ares, L. Extensive-stage small-cell lung cancer: first-line and second-line treatment options. J. Clin. Oncol. 40, 671–681 (2022). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
