

Basal-epithelial subpopulations underlie and predict chemotherapy resistance in triple-negative breast cancer
- PMID: 38480932
- PMCID: PMC11018633
- DOI: 10.1038/s44321-024-00050-0
Basal-epithelial subpopulations underlie and predict chemotherapy resistance in triple-negative breast cancer
Abstract
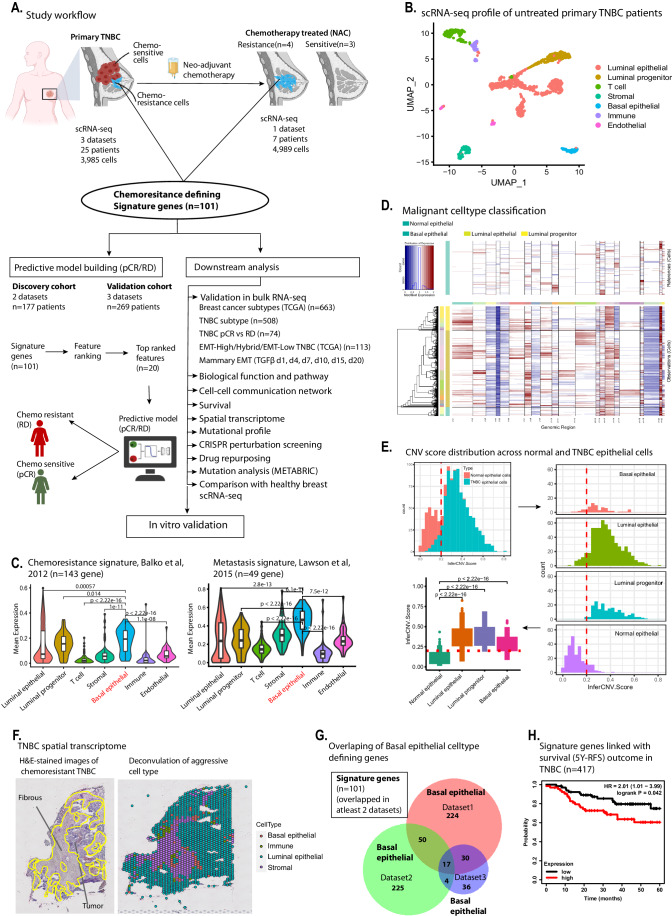
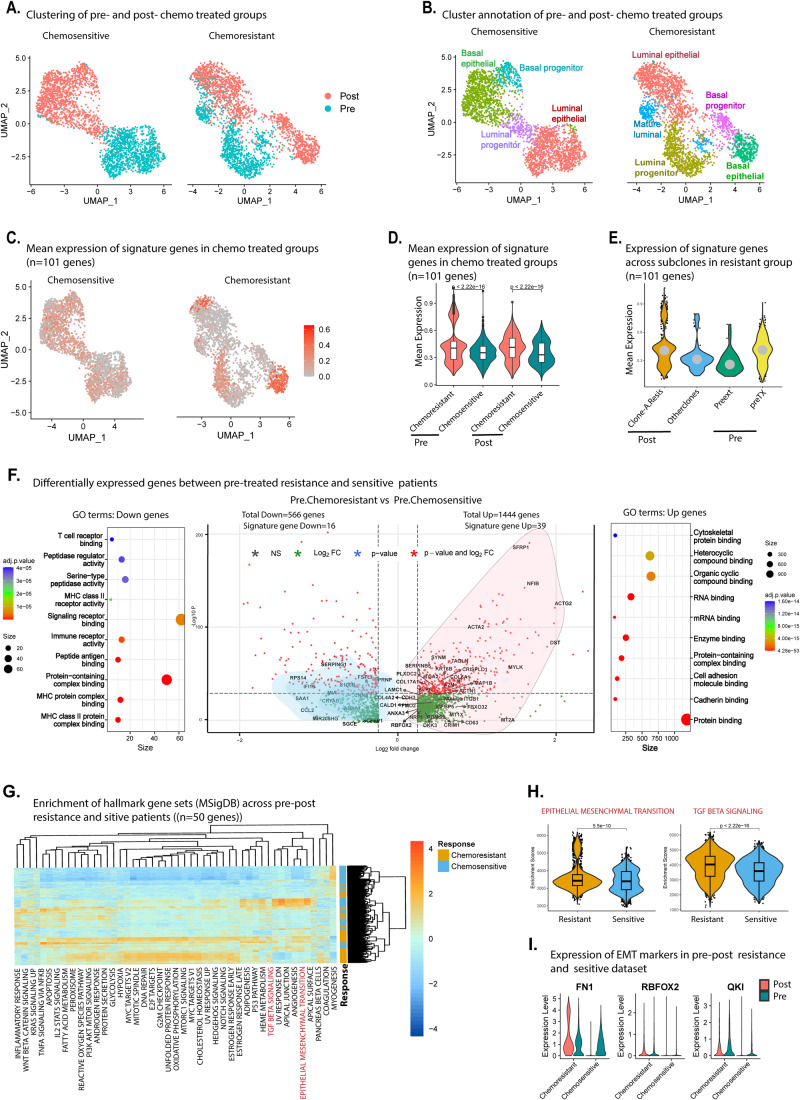
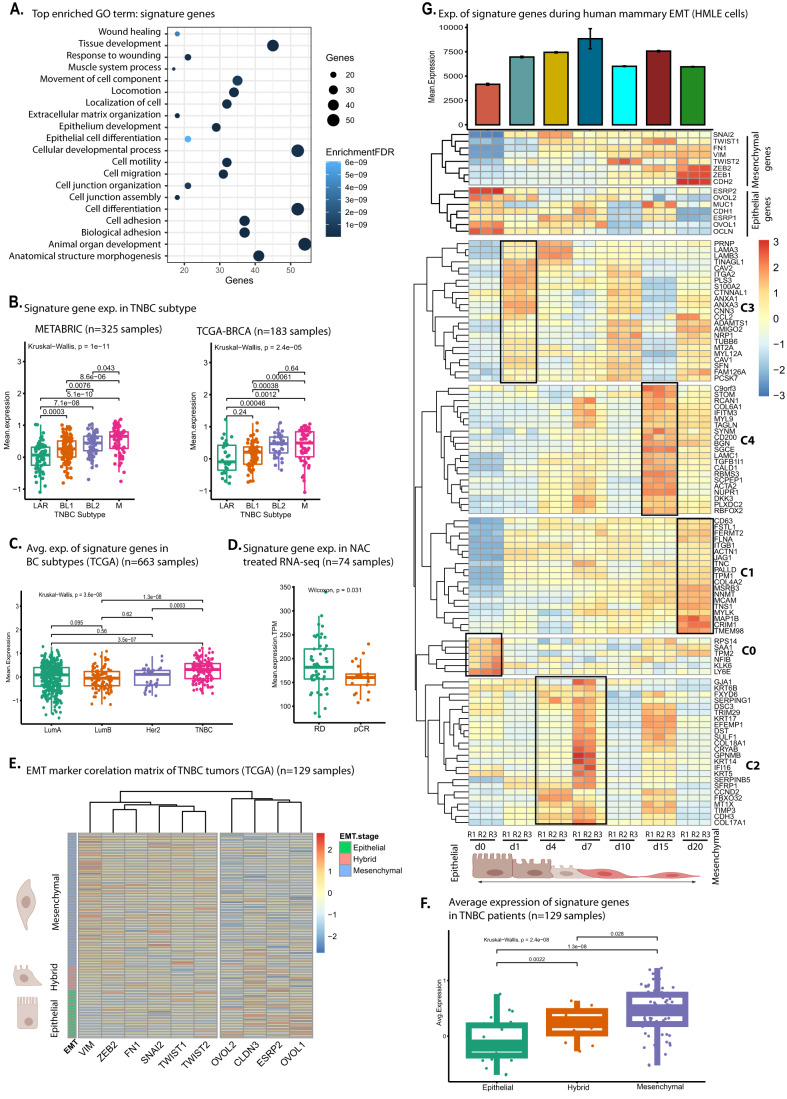
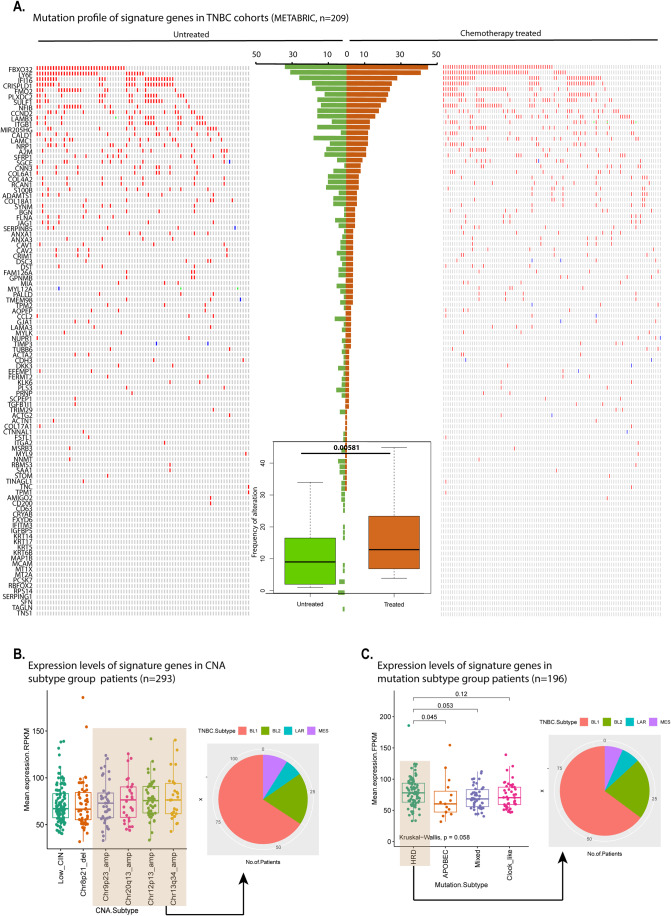
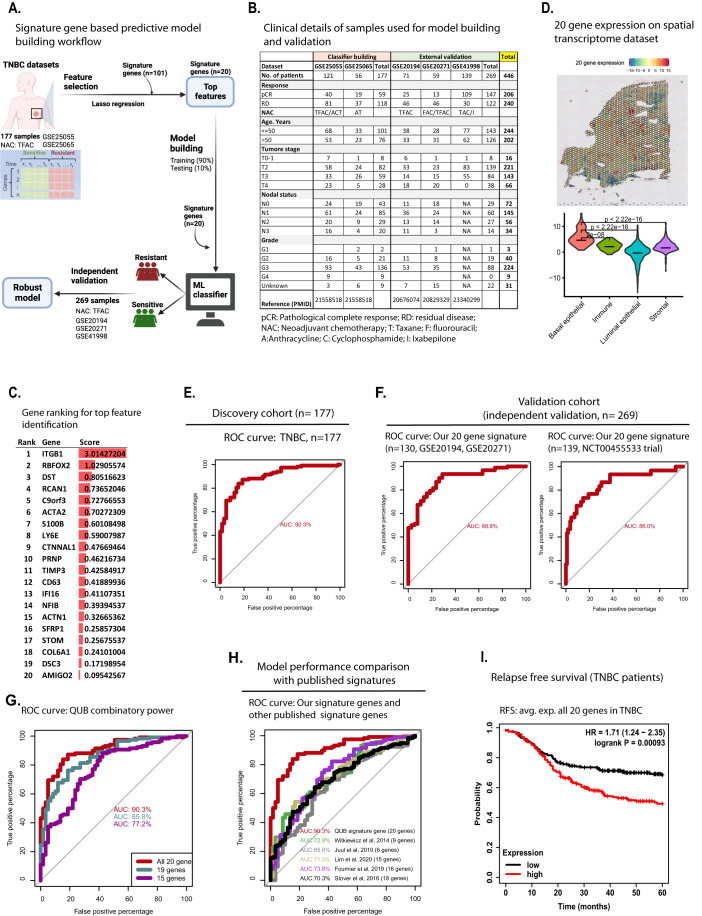
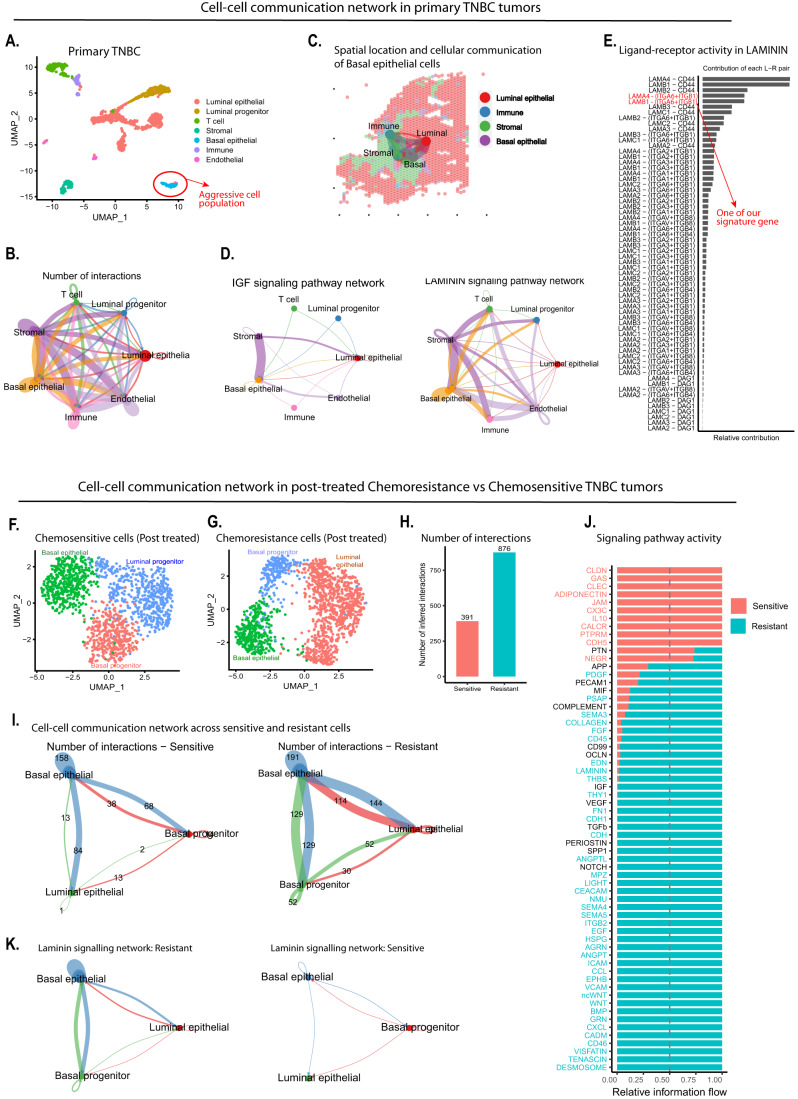
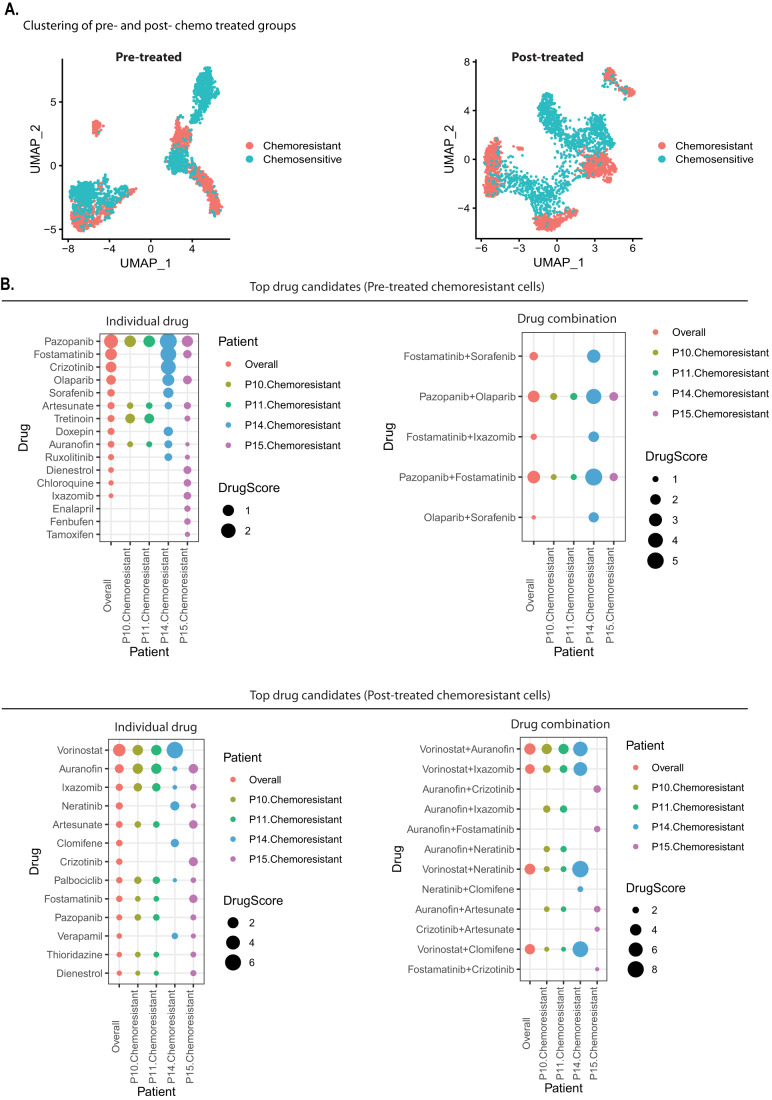
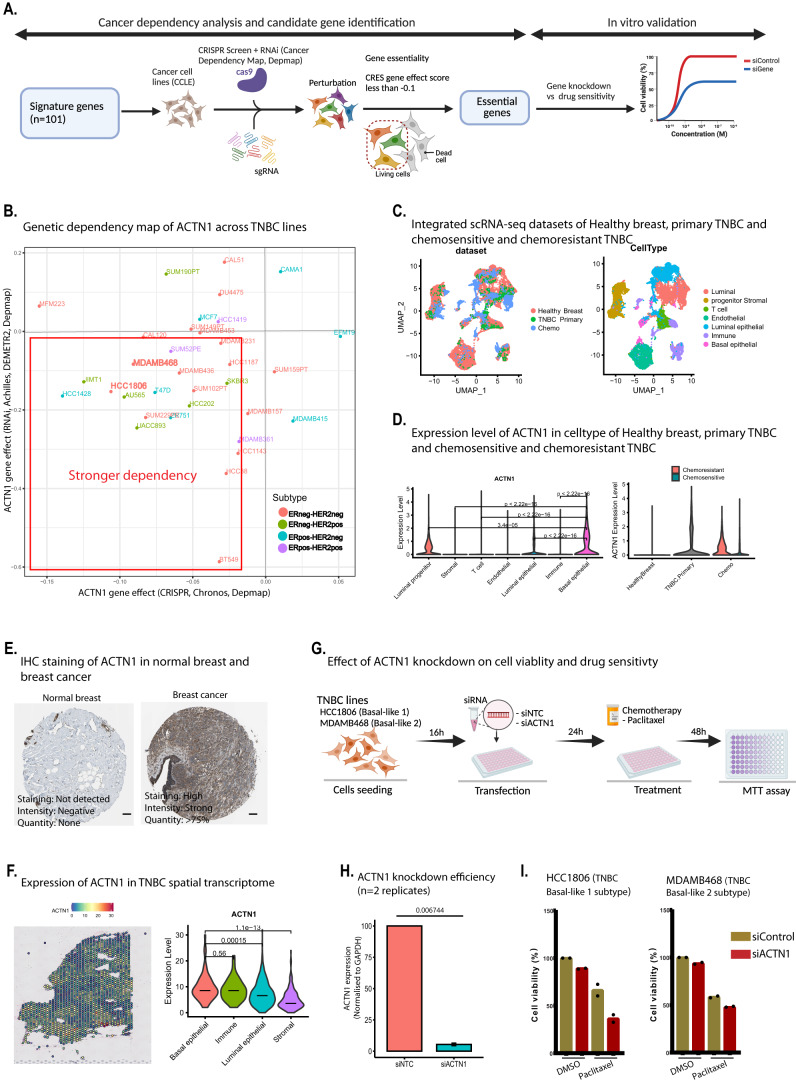
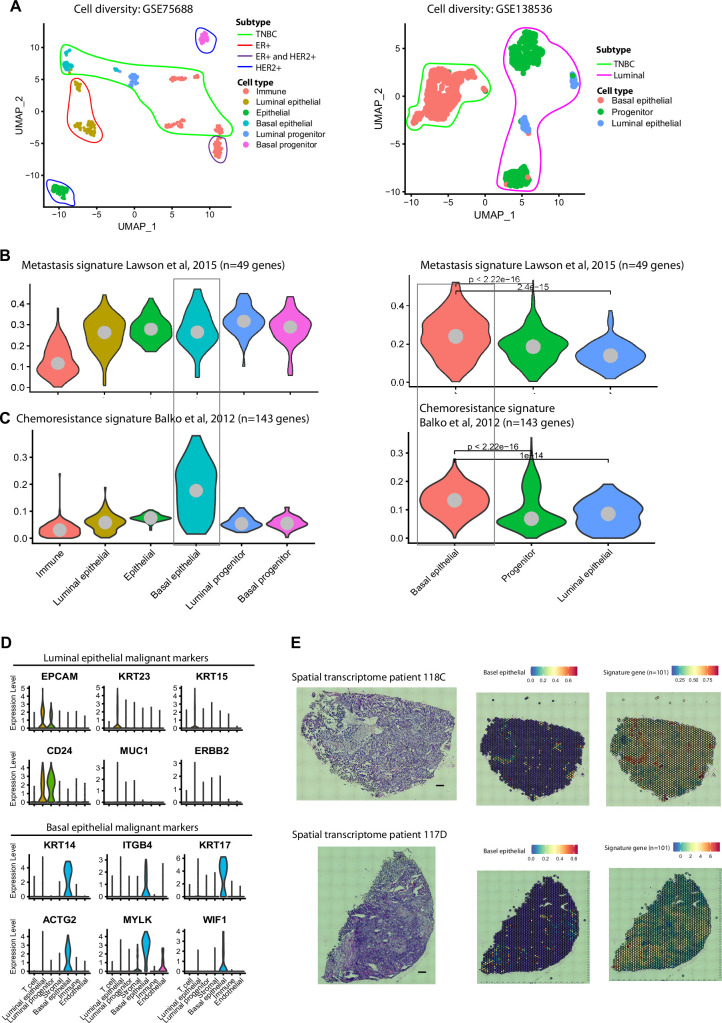
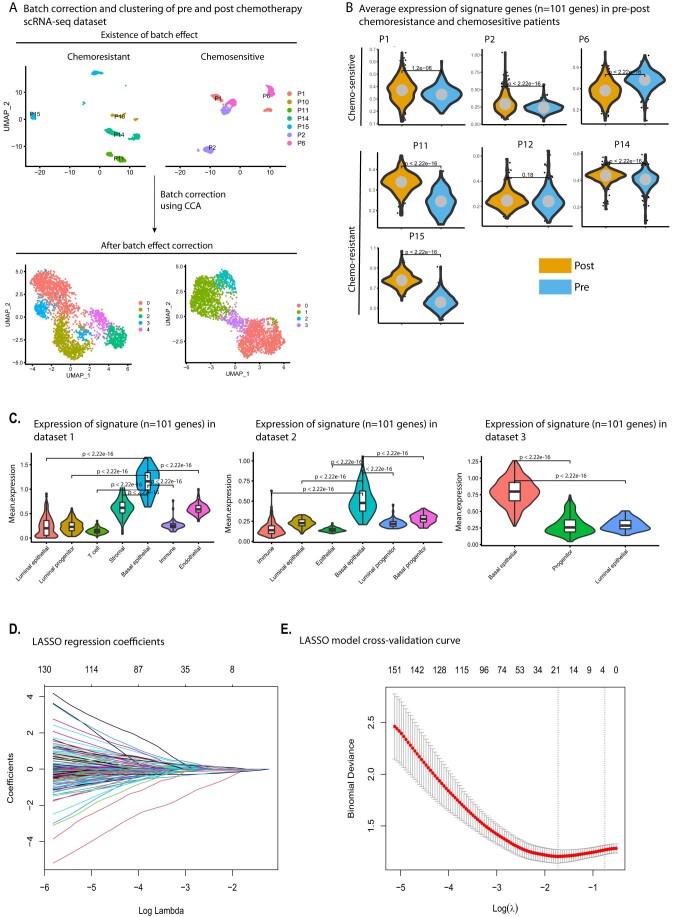
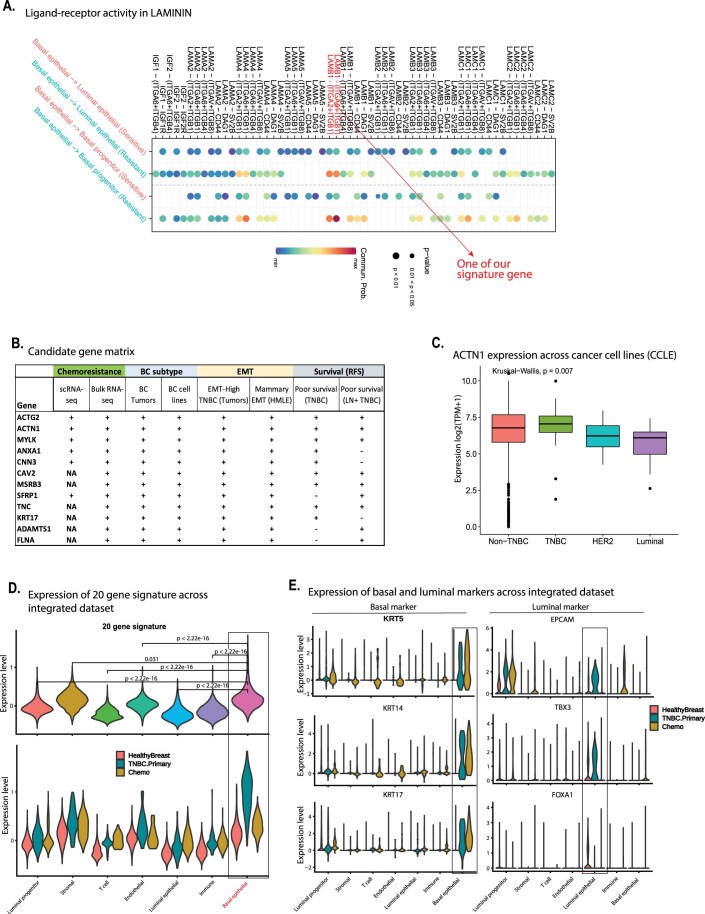
Triple-negative breast cancer (TNBC) is the most aggressive breast cancer subtype, characterized by extensive intratumoral heterogeneity, high metastasis, and chemoresistance, leading to poor clinical outcomes. Despite progress, the mechanistic basis of these aggressive behaviors remains poorly understood. Using single-cell and spatial transcriptome analysis, here we discovered basal epithelial subpopulations located within the stroma that exhibit chemoresistance characteristics. The subpopulations are defined by distinct signature genes that show a frequent gain in copy number and exhibit an activated epithelial-to-mesenchymal transition program. A subset of these genes can accurately predict chemotherapy response and are associated with poor prognosis. Interestingly, among these genes, elevated ITGB1 participates in enhancing intercellular signaling while ACTN1 confers a survival advantage to foster chemoresistance. Furthermore, by subjecting the transcriptional signatures to drug repurposing analysis, we find that chemoresistant tumors may benefit from distinct inhibitors in treatment-naive versus post-NAC patients. These findings shed light on the mechanistic basis of chemoresistance while providing the best-in-class biomarker to predict chemotherapy response and alternate therapeutic avenues for improved management of TNBC patients resistant to chemotherapy.
Keywords: Breast Cancer; EMT; Genomics; Metastasis; Therapy Resistance.
© 2024. The Author(s).
Figures
References
-
- Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez-Farre X, Munoz M, Russnes HG, Helland A, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014;6:514–527. doi: 10.1016/j.celrep.2013.12.041. - DOI - PMC - PubMed
-
- Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, Sanders ME, Granja-Ingram NM, Smith JJ, Meszoely IM, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med. 2012;18:1052–1059. doi: 10.1038/nm.2795. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
