

Classification of likely functional class for ligand binding sites identified from fragment screening
- PMID: 38480979
- PMCID: PMC10937669
- DOI: 10.1038/s42003-024-05970-8
Classification of likely functional class for ligand binding sites identified from fragment screening
Abstract
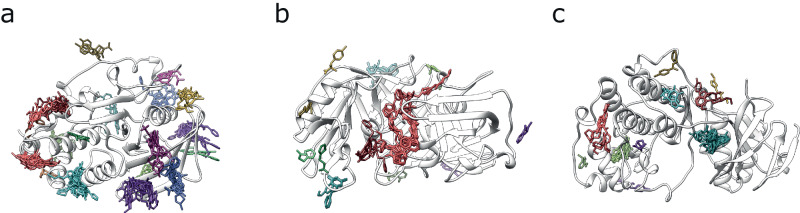
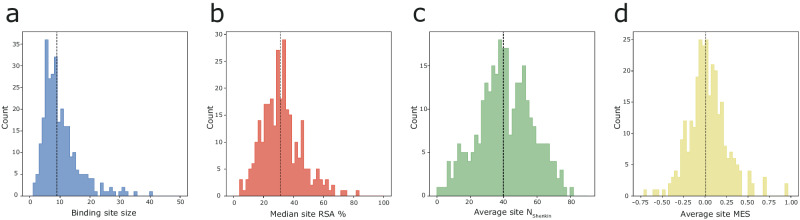
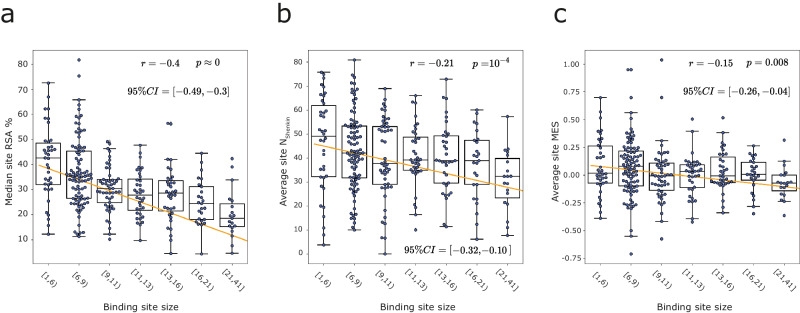
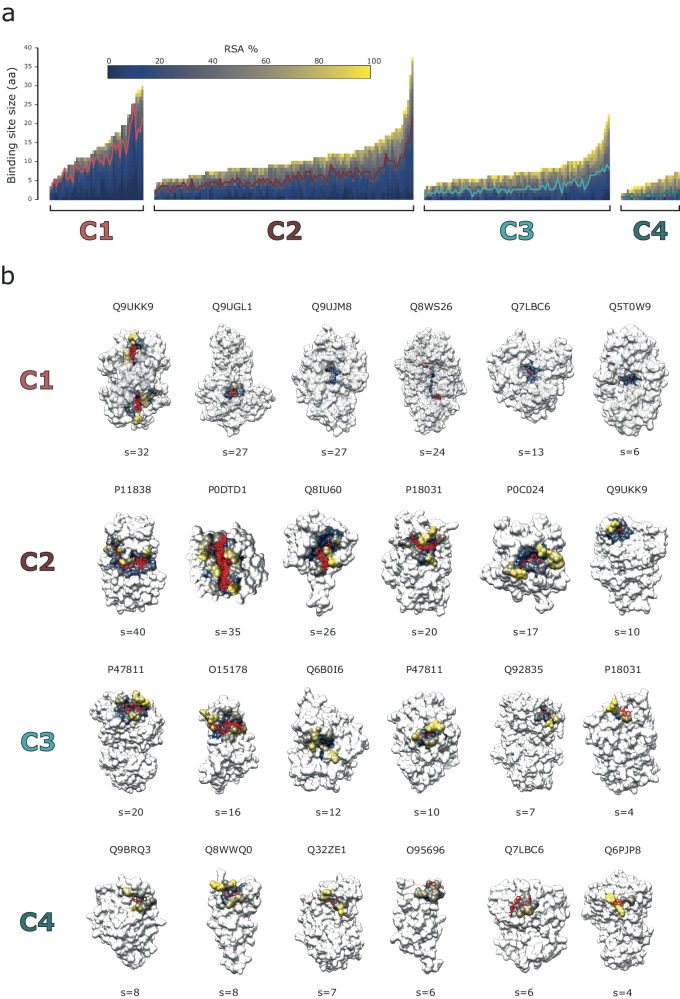
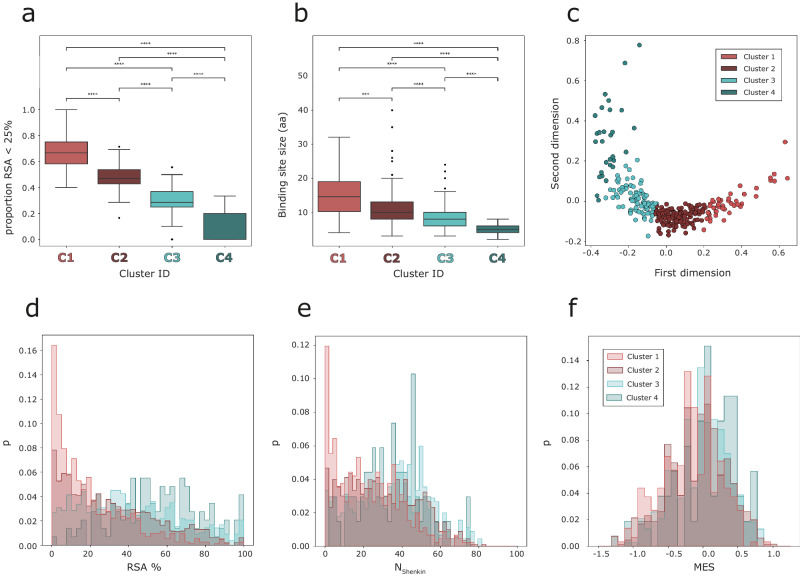
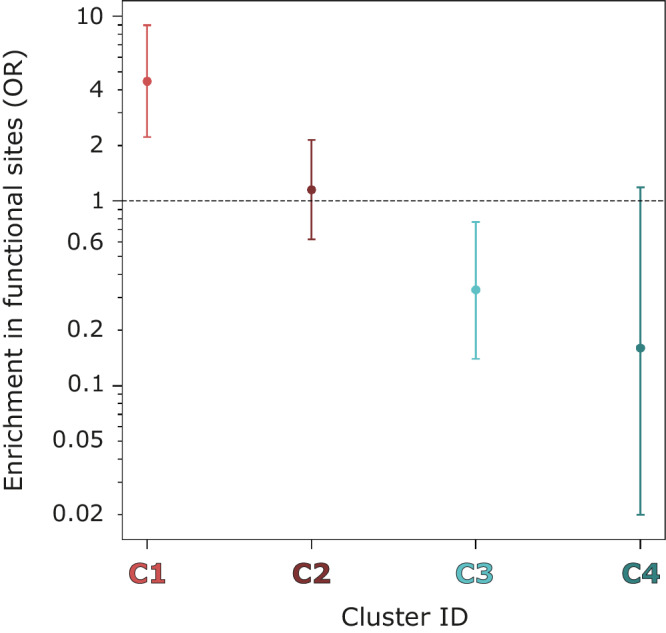
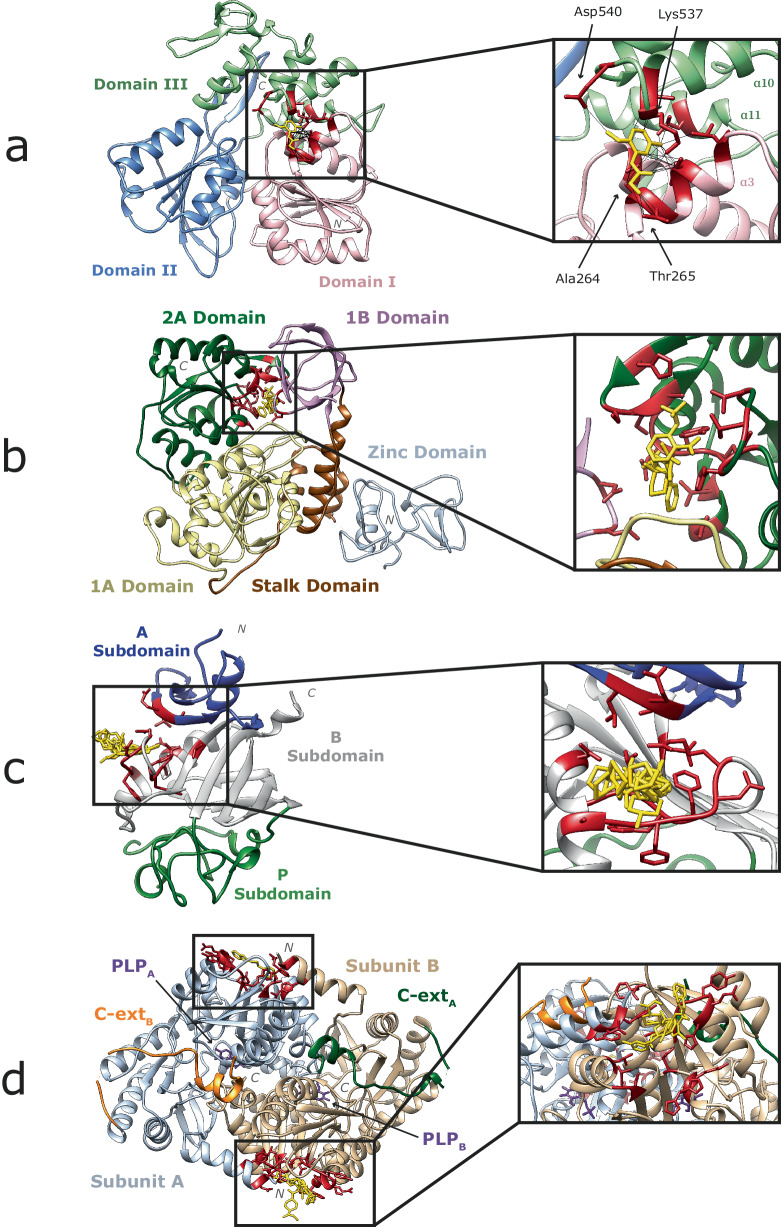
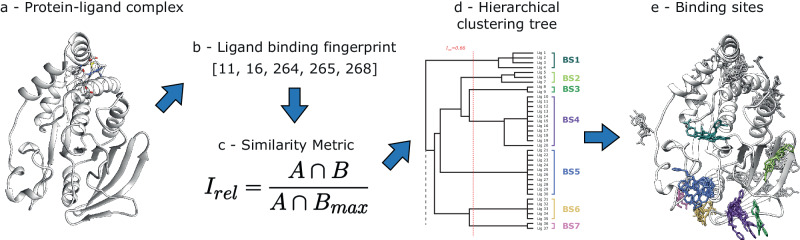
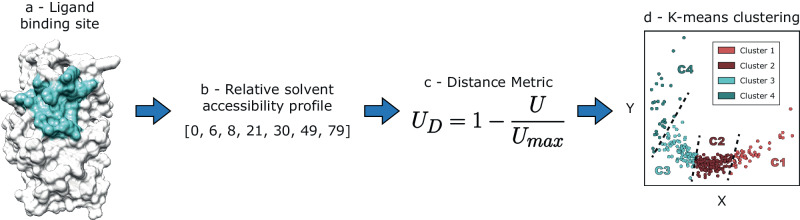
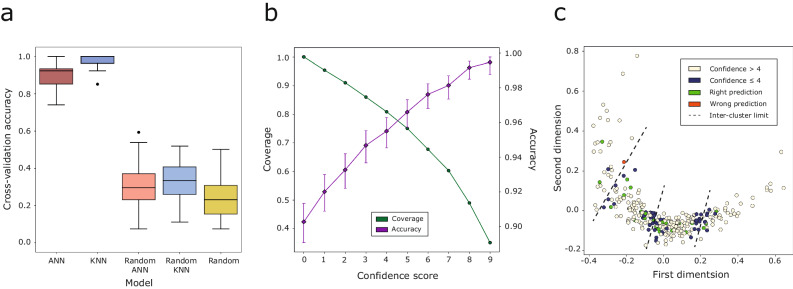
Fragment screening is used to identify binding sites and leads in drug discovery, but it is often unclear which binding sites are functionally important. Here, data from 37 experiments, and 1309 protein structures binding to 1601 ligands were analysed. A method to group ligands by binding sites is introduced and sites clustered according to profiles of relative solvent accessibility. This identified 293 unique ligand binding sites, grouped into four clusters (C1-4). C1 includes larger, buried, conserved, and population missense-depleted sites, enriched in known functional sites. C4 comprises smaller, accessible, divergent, missense-enriched sites, depleted in functional sites. A site in C1 is 28 times more likely to be functional than one in C4. Seventeen sites, which to the best of our knowledge are novel, in 13 proteins are identified as likely to be functionally important with examples from human tenascin and 5-aminolevulinate synthase highlighted. A multi-layer perceptron, and K-nearest neighbours model are presented to predict cluster labels for ligand binding sites with an accuracy of 96% and 100%, respectively, so allowing functional classification of sites for proteins not in this set. Our findings will be of interest to those studying protein-ligand interactions and developing new drugs or function modulators.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
