

De novo-designed transmembrane proteins bind and regulate a cytokine receptor
- PMID: 38480980
- PMCID: PMC11142920
- DOI: 10.1038/s41589-024-01562-z
De novo-designed transmembrane proteins bind and regulate a cytokine receptor
Abstract
Transmembrane (TM) domains as simple as a single span can perform complex biological functions using entirely lipid-embedded chemical features. Computational design has the potential to generate custom tool molecules directly targeting membrane proteins at their functional TM regions. Thus far, designed TM domain-targeting agents have been limited to mimicking the binding modes and motifs of natural TM interaction partners. Here, we demonstrate the design of de novo TM proteins targeting the erythropoietin receptor (EpoR) TM domain in a custom binding topology competitive with receptor homodimerization. The TM proteins expressed in mammalian cells complex with EpoR and inhibit erythropoietin-induced cell proliferation. In vitro, the synthetic TM domain complex outcompetes EpoR homodimerization. Structural characterization reveals that the complex involves the intended amino acids and agrees with our designed molecular model of antiparallel TM helices at 1:1 stoichiometry. Thus, membrane protein TM regions can now be targeted in custom-designed topologies.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
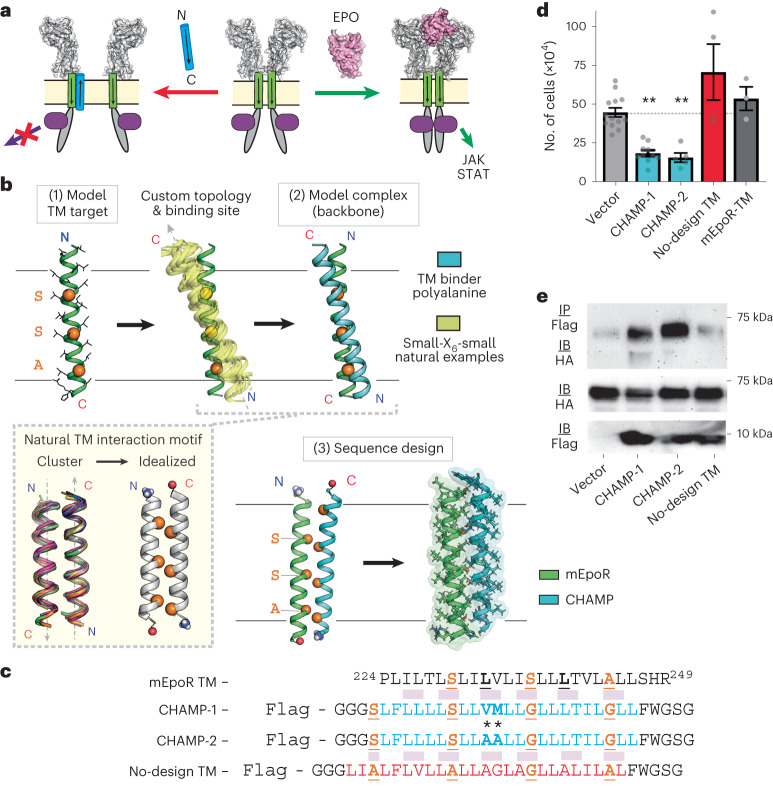

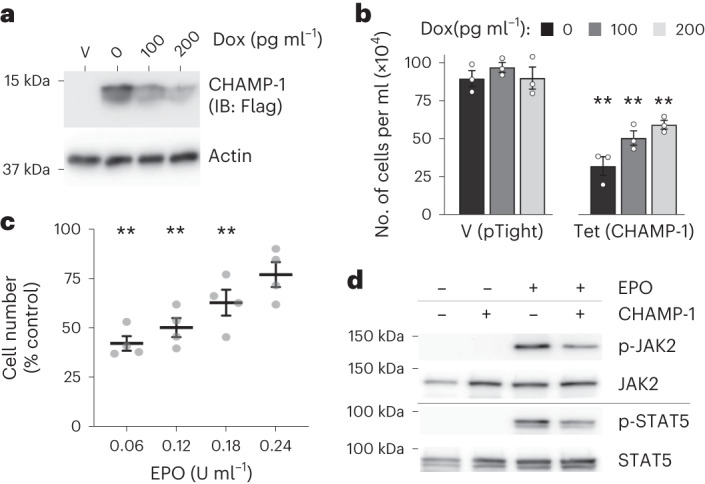

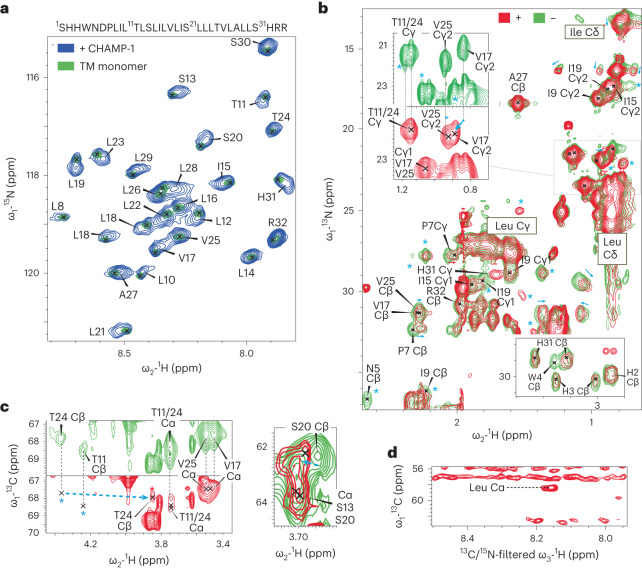
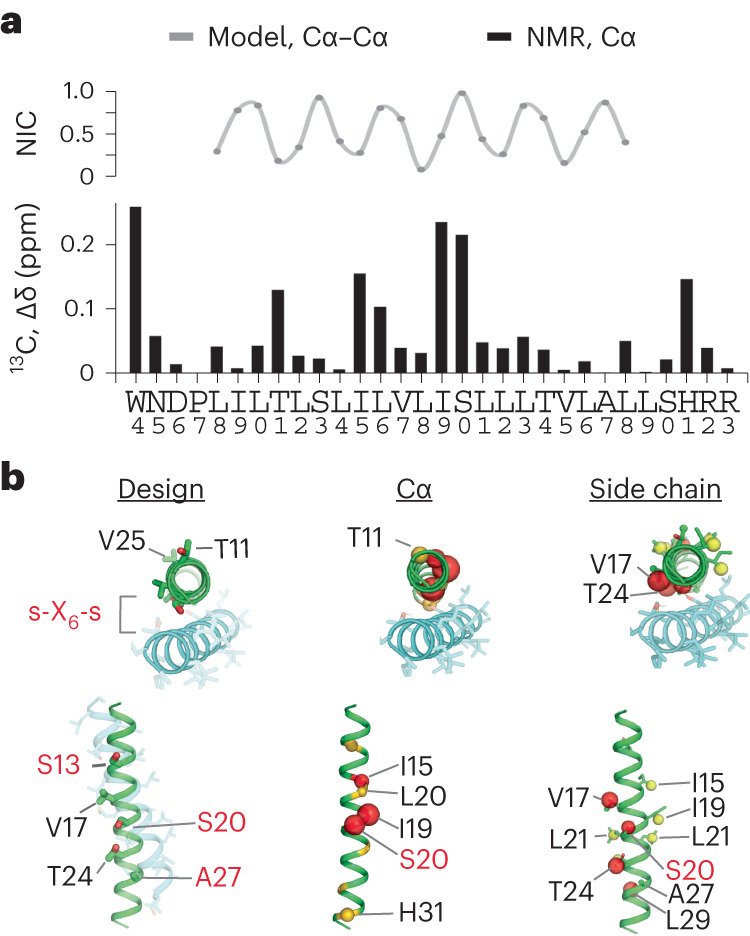










References
Publication types
MeSH terms
Substances
Grants and funding
- R35-122603/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R37 CA037157/CA/NCI NIH HHS/United States
- GM68933/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 CA037157/CA/NCI NIH HHS/United States
- P41 GM068933/GM/NIGMS NIH HHS/United States
