

Inhibition of BRD4 Attenuates ER Stress-induced Renal Ischemic-Reperfusion Injury
- PMID: 38481808
- PMCID: PMC10929187
- DOI: 10.7150/ijbs.83040
Inhibition of BRD4 Attenuates ER Stress-induced Renal Ischemic-Reperfusion Injury
Abstract
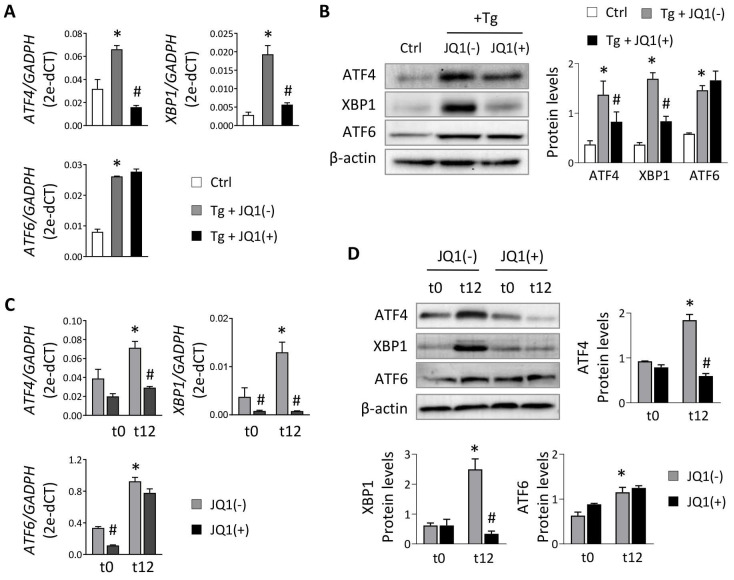
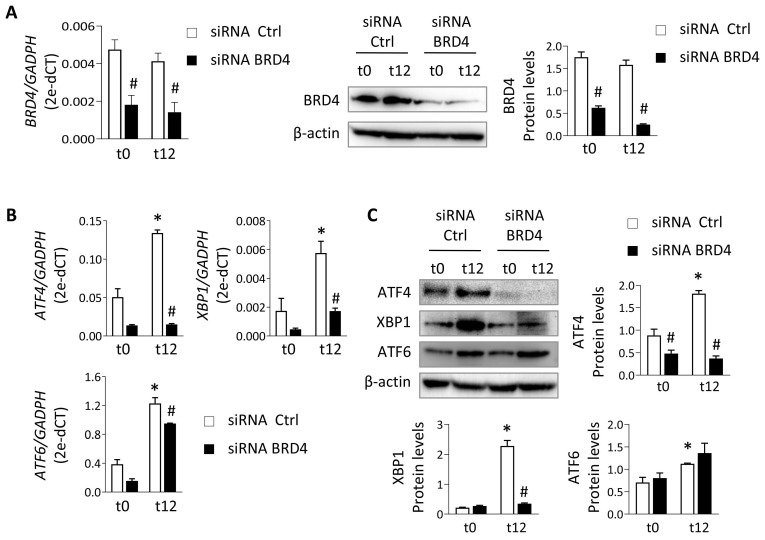
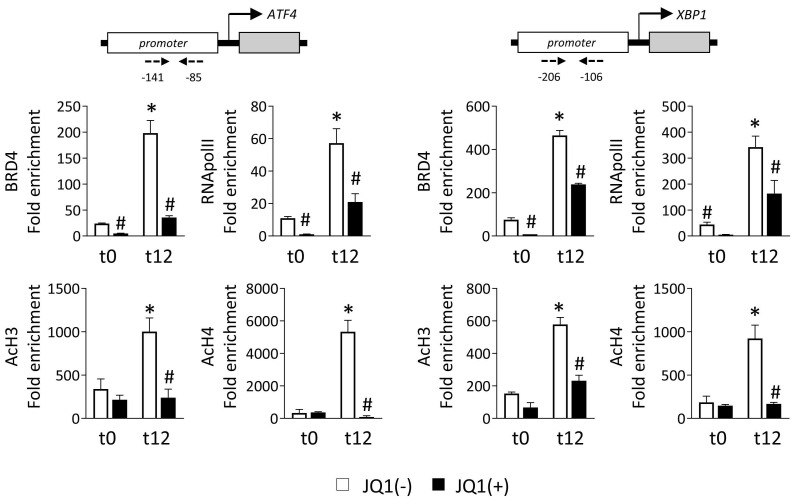
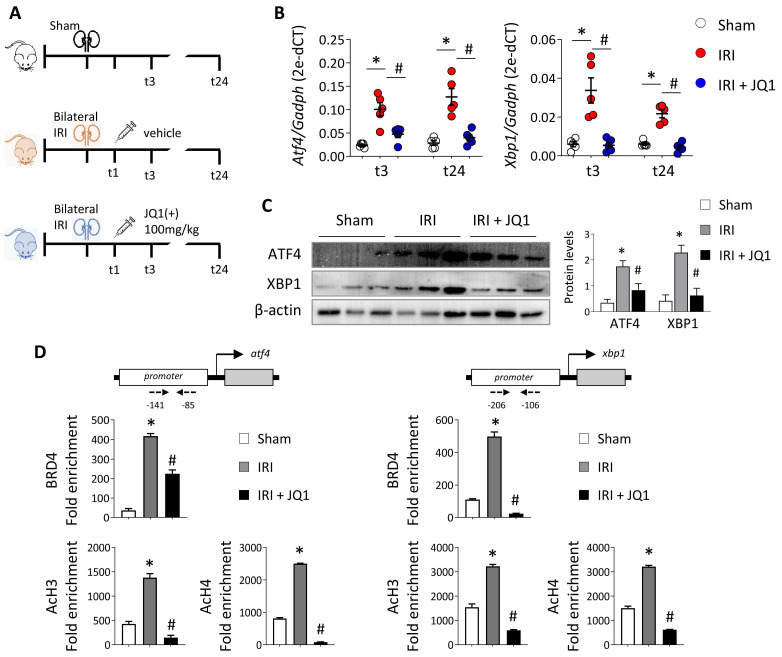
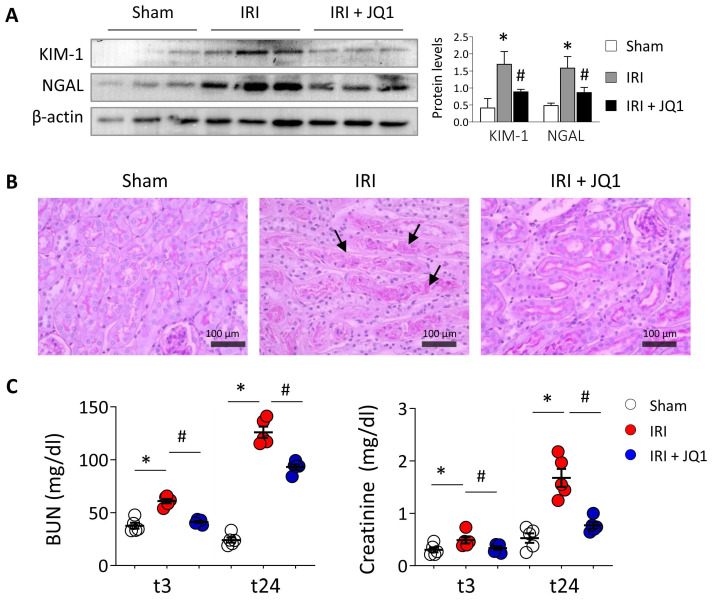
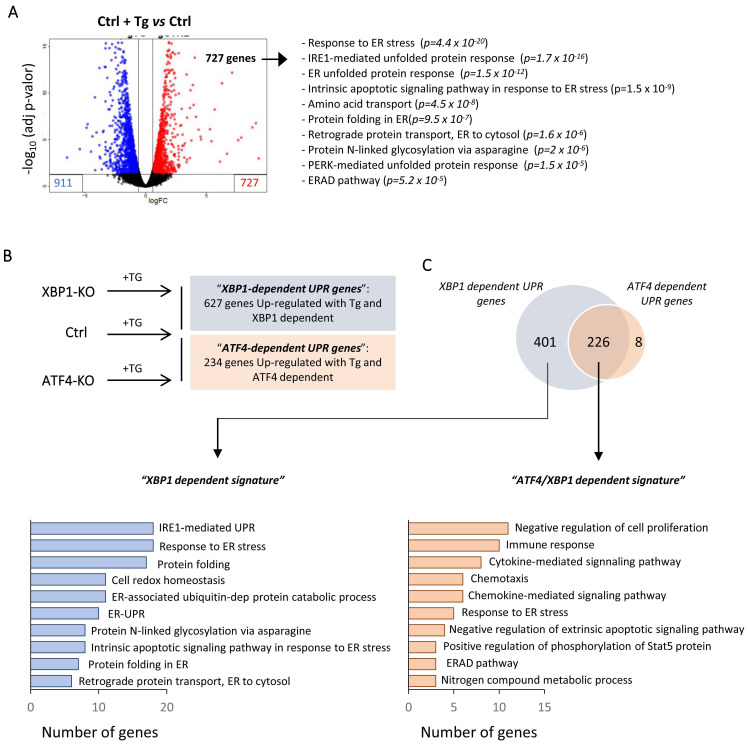
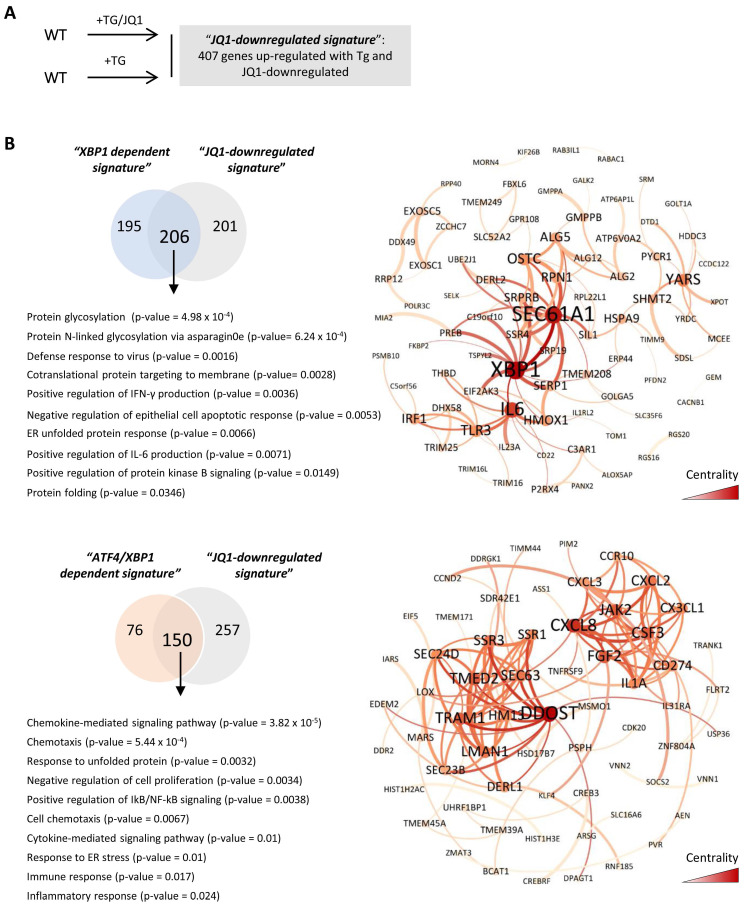
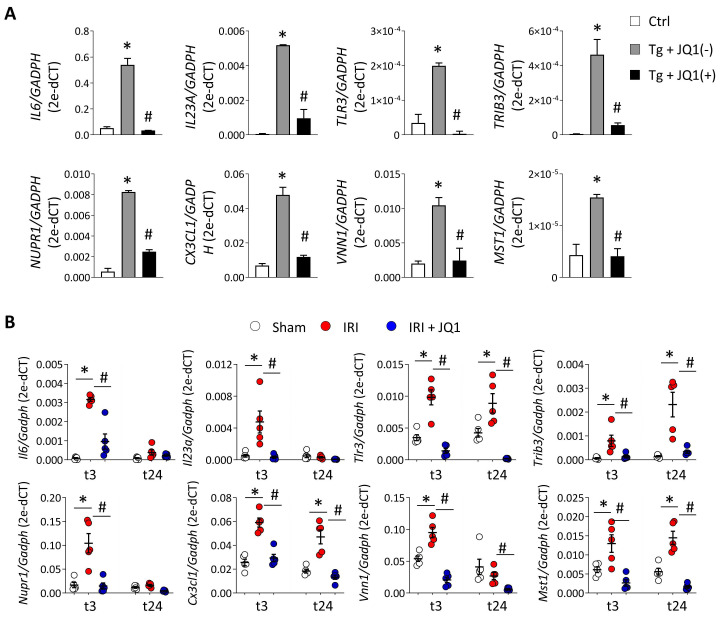
Renal ischemia-reperfusion injury (IRI) leads to endoplasmic reticulum (ER) stress, thereby initiating the unfolded protein response (UPR). When sustained, this response may trigger the inflammation and tubular cell death that acts to aggravate the damage. Here, we show that knockdown of the BET epigenetic reader BRD4 reduces the expression of ATF4 and XBP1 transcription factors under ER stress activation. BRD4 is recruited to the promoter of these highly acetylated genes, initiating gene transcription. Administration of the BET protein inhibitor, JQ1, one hour after renal damage induced by bilateral IRI, reveals reduced expression of ATF4 and XBP1 genes, low KIM-1 and NGAL levels and recovery of the serum creatinine and blood urea nitrogen levels. To determine the molecular pathways regulated by ATF4 and XBP1, we performed stable knockout of both transcription factors using CRISPR-Cas9 and RNA sequencing. The pathways triggered under ER stress were mainly XBP1-dependent, associated with an adaptive UPR, and partially regulated by JQ1. Meanwhile, treatment with JQ1 downmodulated most of the pathways regulated by ATF4 and related to the pathological processes during exacerbated UPR activation. Thus, BRD4 inhibition could be useful for curbing the maladaptive UPR activation mechanisms, thereby ameliorating the progression of renal disease.
Keywords: BRD4; ER stress; Hypoxia; UPR; renal damage.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nature reviews Nephrology. 2017;13:681–96. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
