

Carbonic Anhydrase 3 is required for cardiac repair post myocardial infarction via Smad7-Smad2/3 signaling pathway
- PMID: 38481818
- PMCID: PMC10929199
- DOI: 10.7150/ijbs.91396
Carbonic Anhydrase 3 is required for cardiac repair post myocardial infarction via Smad7-Smad2/3 signaling pathway
Abstract
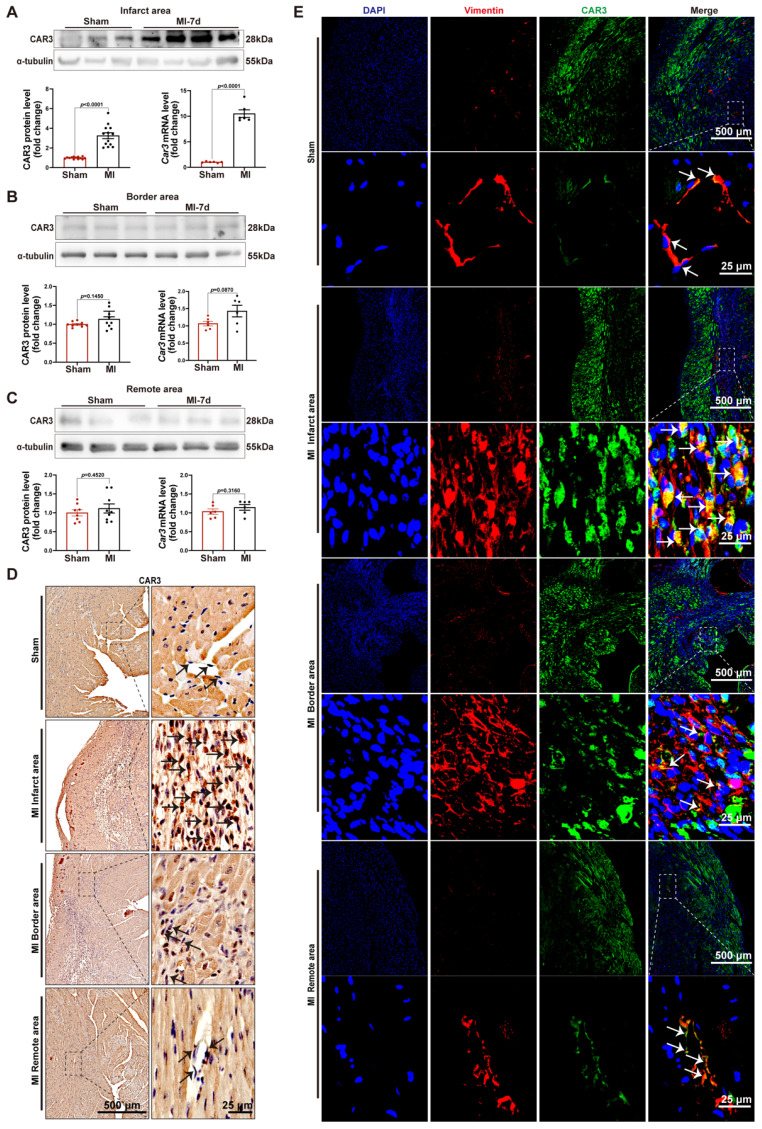
Appropriate fibrosis is required to prevent subsequent adverse remodeling and heart failure post myocardial infarction (MI), and cardiac fibroblasts (CFs) play a critical role during the process. Carbonic anhydrase 3 (CAR3) is an important mediator in multiple biological processes besides its CO2 hydration activity; however, the role and underlying mechanism of CAR3 on cardiac repair post MI injury remains unknown. Here, we found that CAR3 expression was up-regulated in cardiac tissue in infarct area at the reparative phase of MI, with a peak at 7 days post MI. The upregulation was detected mainly on fibroblast instead of cardiomyocyte, and primary cardiac fibroblasts treated with TGF-β1 recaptured our observation. While CAR3 deficiency leads to weakened collagen density, enlarged infarct size and aggravated cardiac dysfunction post-MI. In fibroblast, we observed that CAR3 deficiency restrains collagen synthesis, cell migration and gel contraction of cardiac fibroblasts, whereas overexpression of CAR3 in CFs improves wound healing and cardiac fibroblast activation. Mechanistically, CAR3 stabilizes Smad7 protein via modulating its acetylation, which dampens phosphorylation of Smad2 and Smad3, thus inhibiting fibroblast transformation. In contrast, inhibition of Smad7 acetylation with C646 blunts CAR3 deficiency induced suppression of fibroblast activation and impaired cardiac healing. Our data demonstrate a protective role of CAR3 in cardiac wound repair post MI via promoting fibroblasts activation through Smad7-TGF-β/Smad2/3 signaling pathway.
Keywords: CAR3; Smad7 acetylation; cardiac repair; fibroblast activation; myocardial infarction.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures








Similar articles
-
Exercise Training Alleviates Cardiac Fibrosis through Increasing Fibroblast Growth Factor 21 and Regulating TGF-β1-Smad2/3-MMP2/9 Signaling in Mice with Myocardial Infarction.Int J Mol Sci. 2021 Nov 15;22(22):12341. doi: 10.3390/ijms222212341. Int J Mol Sci. 2021. PMID: 34830222 Free PMC article.
-
Lnc-Ang362 is a pro-fibrotic long non-coding RNA promoting cardiac fibrosis after myocardial infarction by suppressing Smad7.Arch Biochem Biophys. 2020 May 30;685:108354. doi: 10.1016/j.abb.2020.108354. Epub 2020 Mar 30. Arch Biochem Biophys. 2020. PMID: 32240638
-
Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7.Cell Physiol Biochem. 2017;42(6):2207-2219. doi: 10.1159/000479995. Epub 2017 Aug 16. Cell Physiol Biochem. 2017. PMID: 28817807
-
Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction.Matrix Biol. 2020 Sep;91-92:136-151. doi: 10.1016/j.matbio.2020.03.007. Epub 2020 Mar 21. Matrix Biol. 2020. PMID: 32209358 Free PMC article. Review.
-
Smad-dependent pathways in the infarcted and failing heart.Curr Opin Pharmacol. 2022 Jun;64:102207. doi: 10.1016/j.coph.2022.102207. Epub 2022 Mar 31. Curr Opin Pharmacol. 2022. PMID: 35367786 Free PMC article. Review.
Cited by
-
Active ingredients of traditional Chinese medicine inhibit NOD-like receptor protein 3 inflammasome: a novel strategy for preventing and treating heart failure.Front Immunol. 2025 Jan 24;16:1520482. doi: 10.3389/fimmu.2025.1520482. eCollection 2025. Front Immunol. 2025. PMID: 39925805 Free PMC article. Review.
-
GEO combined with quantitative protein trait loci identify causative proteins in hypertrophic cardiomyopathy.ESC Heart Fail. 2025 Aug;12(4):2827-2833. doi: 10.1002/ehf2.15287. Epub 2025 May 9. ESC Heart Fail. 2025. PMID: 40348582 Free PMC article.
-
SMAD7: riding on fibrosis-limiting routes and beyond.EMBO Mol Med. 2025 Jul 31. doi: 10.1038/s44321-025-00283-7. Online ahead of print. EMBO Mol Med. 2025. PMID: 40745212 Review.
References
-
- Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS. et al. Heart Disease and Stroke Statistics-2022 Update: A Report From the American Heart Association. Circulation. 2022;145:e153–e639. - PubMed
-
- Vaduganathan M, Mensah GA, Turco JV, Fuster V, Roth GA. The Global Burden of Cardiovascular Diseases and Risk: A Compass for Future Health. J Am Coll Cardiol. 2022;80:2361–2371. - PubMed
-
- Mensah GA, Roth GA, Fuster V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J Am Coll Cardiol. 2019;74:2529–2532. - PubMed
-
- Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G. et al. Optimized Treatment of ST-Elevation Myocardial Infarction. Circ Res. 2019;125:245–258. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
