

Decoding the tumor microenvironment and molecular mechanism: unraveling cervical cancer subpopulations and prognostic signatures through scRNA-Seq and bulk RNA-seq analyses
- PMID: 38482016
- PMCID: PMC10933018
- DOI: 10.3389/fimmu.2024.1351287
Decoding the tumor microenvironment and molecular mechanism: unraveling cervical cancer subpopulations and prognostic signatures through scRNA-Seq and bulk RNA-seq analyses
Abstract
Background: Cervical carcinoma (CC) represents a prevalent gynecological neoplasm, with a discernible rise in prevalence among younger cohorts observed in recent years. Nonetheless, the intrinsic cellular heterogeneity of CC remains inadequately investigated.
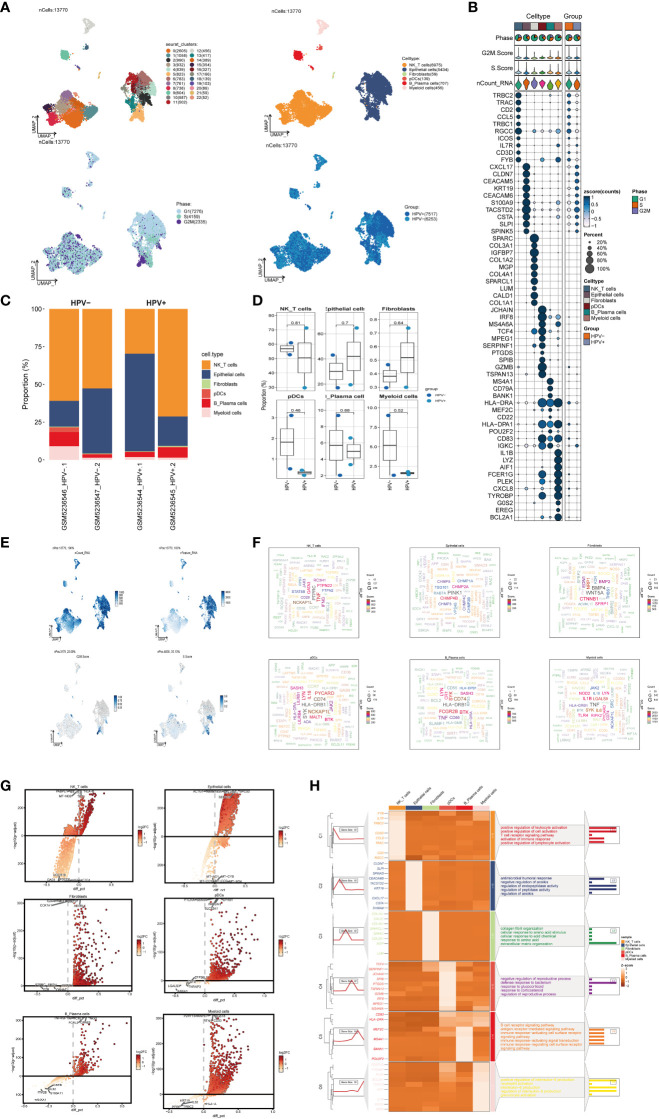
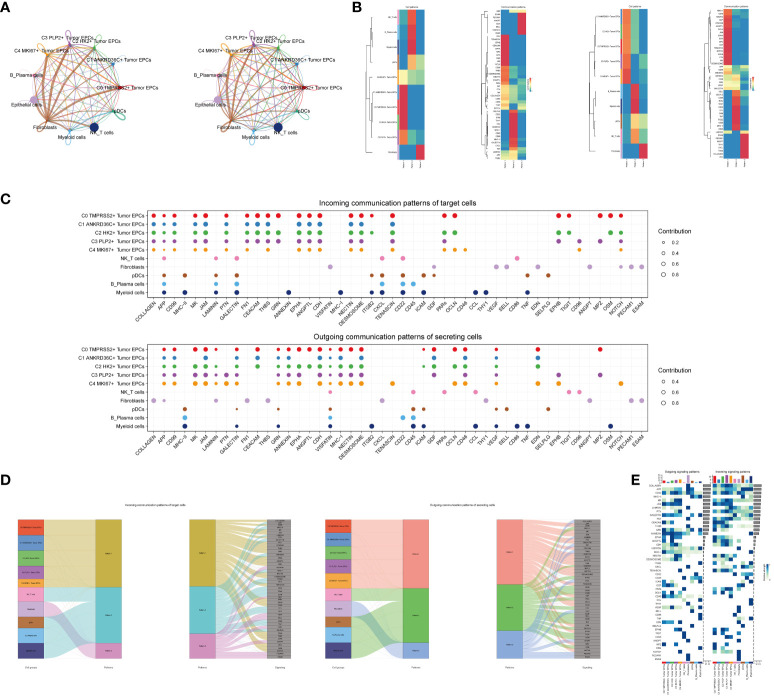
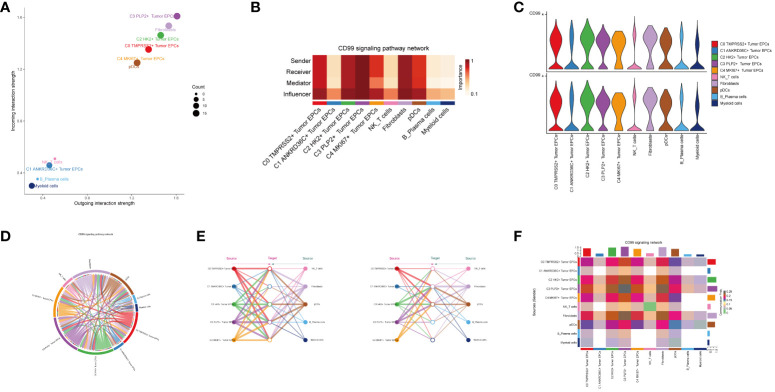
Methods: We utilized single-cell RNA sequencing (scRNA-seq) transcriptomic analysis to scrutinize the tumor epithelial cells derived from four specimens of cervical carcinoma (CC) patients. This method enabled the identification of pivotal subpopulations of tumor epithelial cells and elucidation of their contributions to CC progression. Subsequently, we assessed the influence of associated molecules in bulk RNA sequencing (Bulk RNA-seq) cohorts and performed cellular experiments for validation purposes.
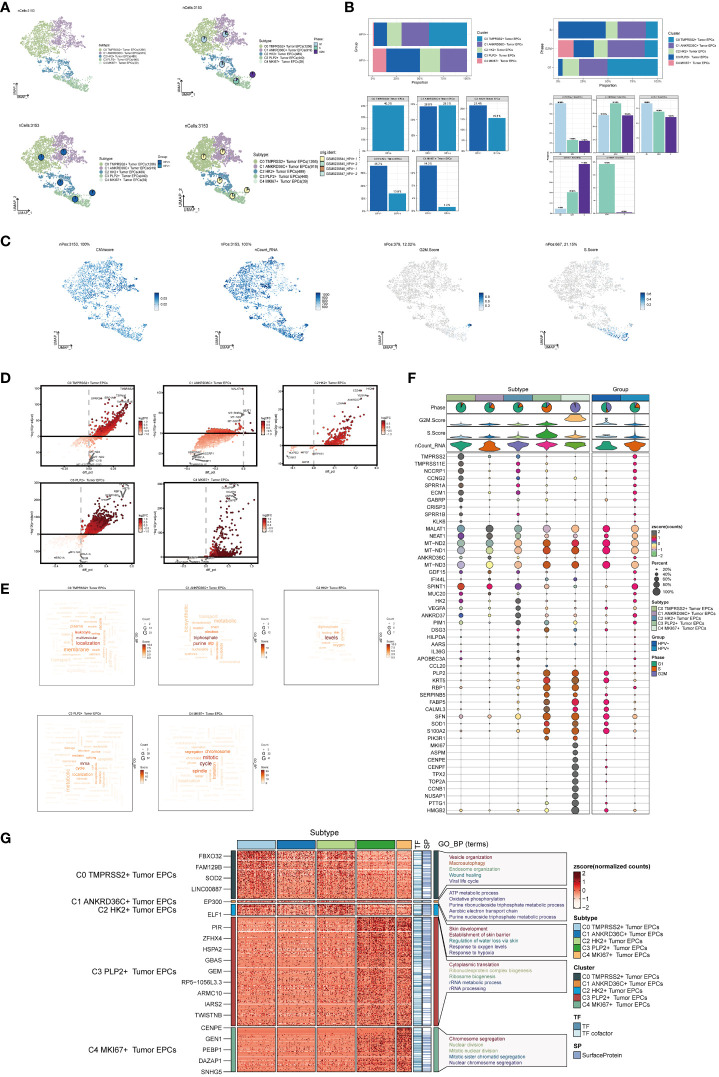
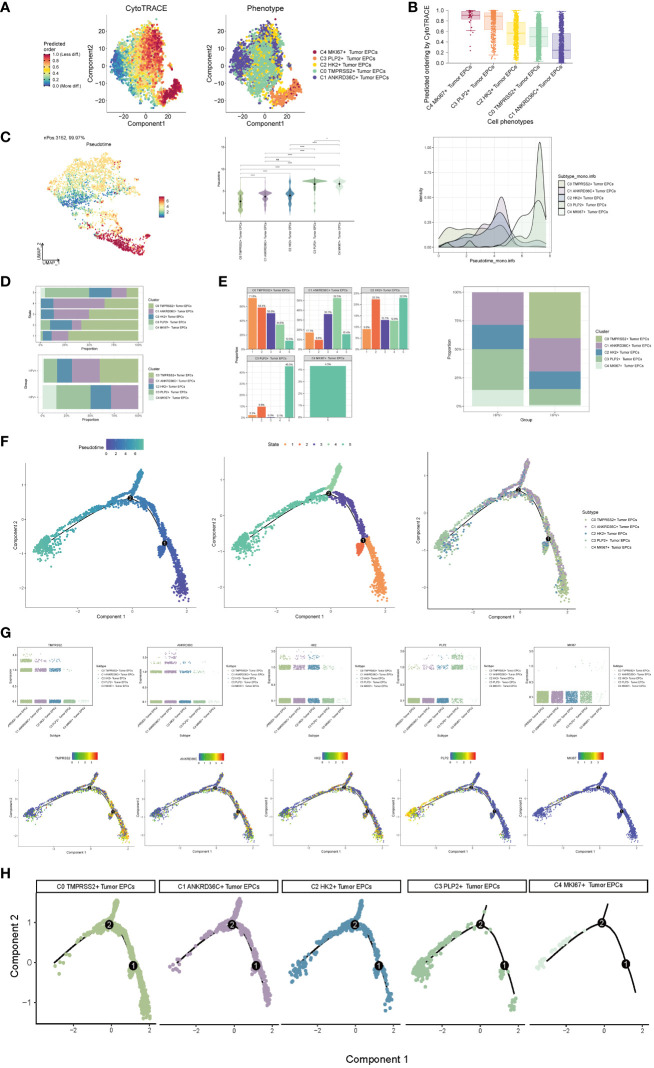
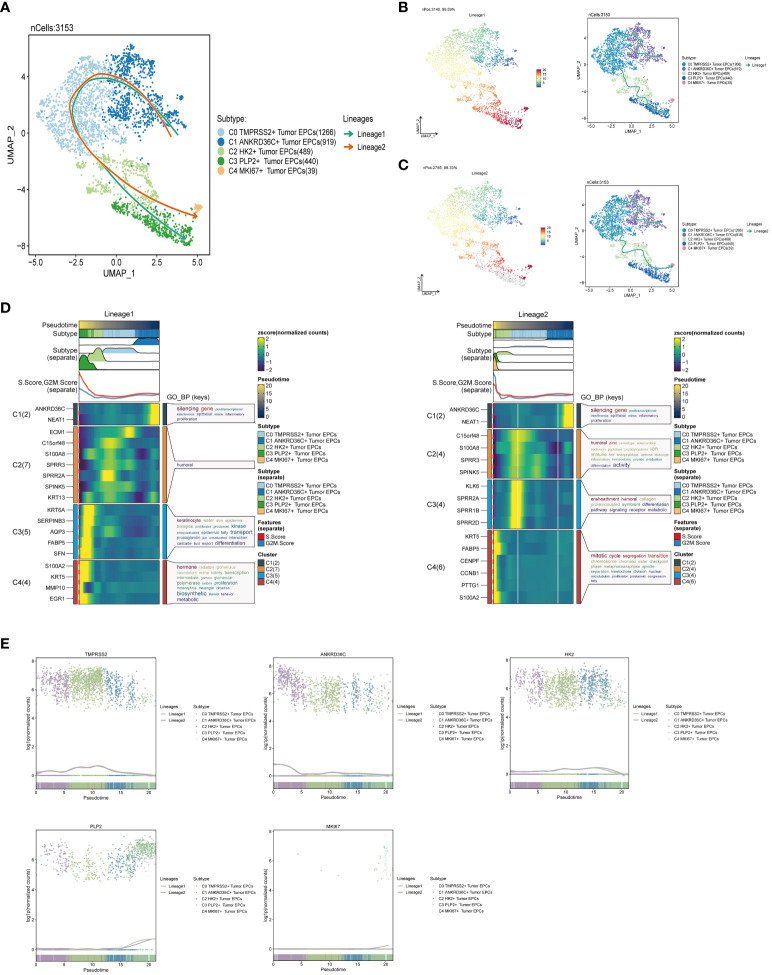
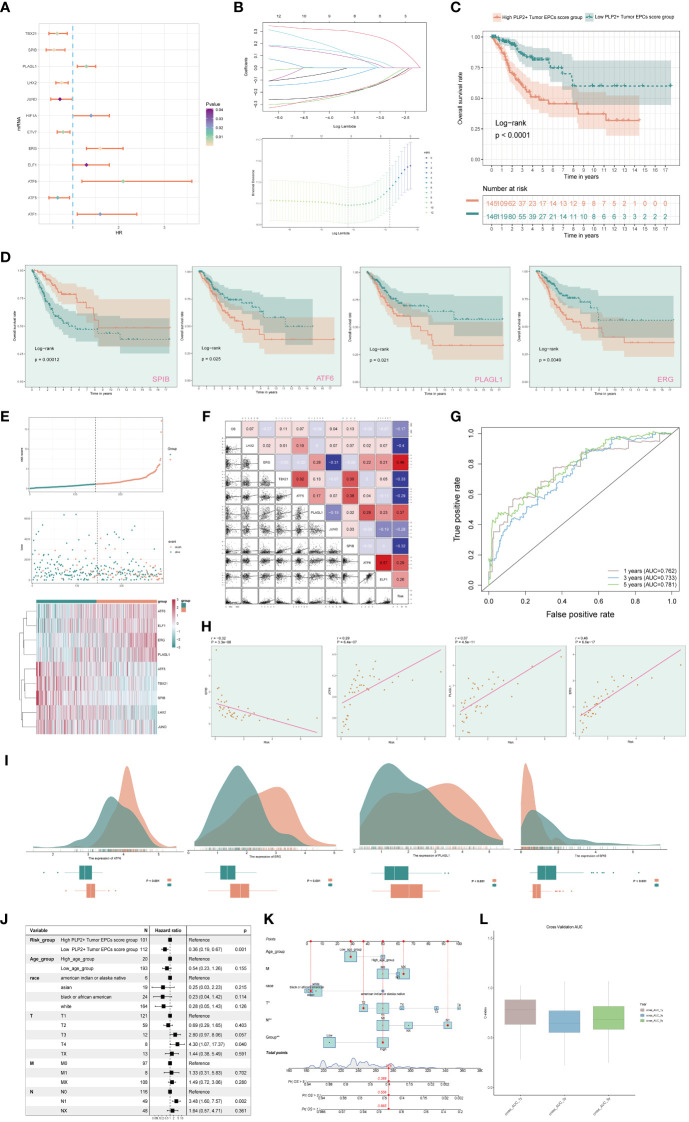
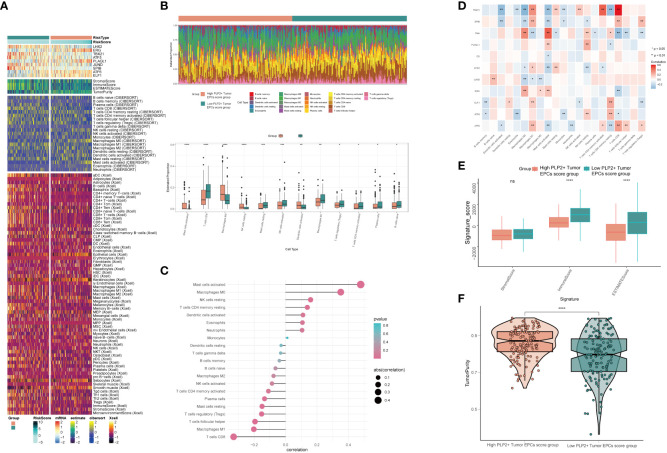
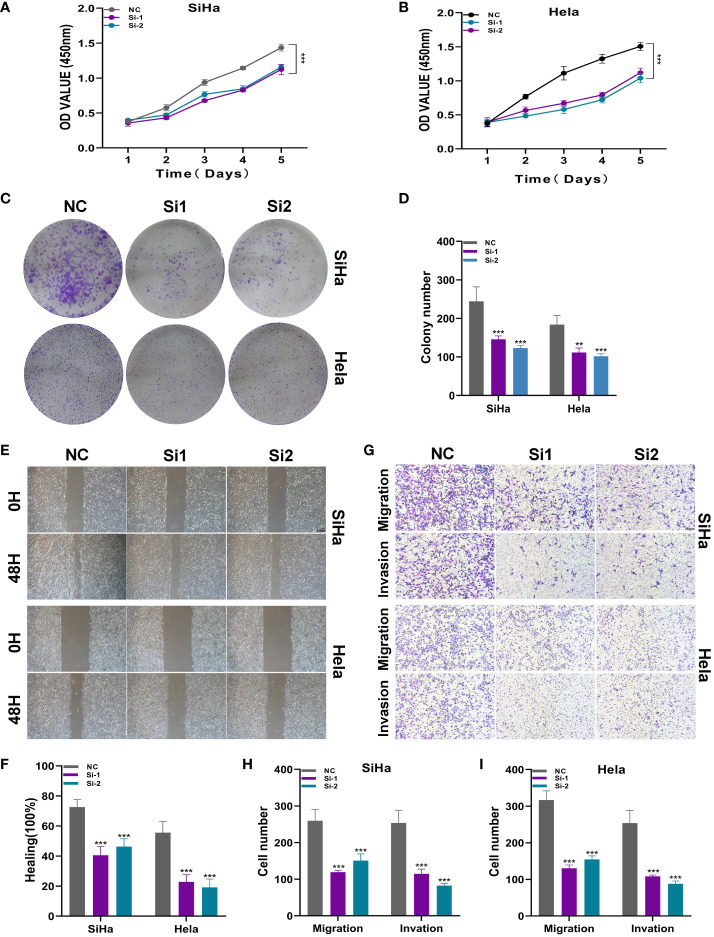
Results: Through our analysis, we have discerned C3 PLP2+ Tumor Epithelial Progenitor Cells as a noteworthy subpopulation in cervical carcinoma (CC), exerting a pivotal influence on the differentiation and progression of CC. We have established an independent prognostic indicator-the PLP2+ Tumor EPCs score. By stratifying patients into high and low score groups based on the median score, we have observed that the high-score group exhibits diminished survival rates compared to the low-score group. The correlations observed between these groups and immune infiltration, enriched pathways, single-nucleotide polymorphisms (SNPs), drug sensitivity, among other factors, further underscore their impact on CC prognosis. Cellular experiments have validated the significant impact of ATF6 on the proliferation and migration of CC cell lines.
Conclusion: This study enriches our comprehension of the determinants shaping the progression of CC, elevates cognizance of the tumor microenvironment in CC, and offers valuable insights for prospective CC therapies. These discoveries contribute to the refinement of CC diagnostics and the formulation of optimal therapeutic approaches.
Keywords: PLP2+ Tumor EPCs; ScRNA-seq; bulk RNA-seq; cervical cancer; experiment validation.
Copyright © 2024 Lin, Li, Shi, Cao, Zhu, Dang, Sheng, Fan, Yang and Wu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous