

MRE11A: a novel negative regulator of human DNA mismatch repair
- PMID: 38486171
- PMCID: PMC10938699
- DOI: 10.1186/s11658-024-00547-z
MRE11A: a novel negative regulator of human DNA mismatch repair
Abstract
Background: DNA mismatch repair (MMR) is a highly conserved pathway that corrects DNA replication errors, the loss of which is attributed to the development of various types of cancers. Although well characterized, MMR factors remain to be identified. As a 3'-5' exonuclease and endonuclease, meiotic recombination 11 homolog A (MRE11A) is implicated in multiple DNA repair pathways. However, the role of MRE11A in MMR is unclear.
Methods: Initially, short-term and long-term survival assays were used to measure the cells' sensitivity to N-methyl-N'-nitro-N-nitrosoguanidine (MNNG). Meanwhile, the level of apoptosis was also determined by flow cytometry after MNNG treatment. Western blotting and immunofluorescence assays were used to evaluate the DNA damage within one cell cycle after MNNG treatment. Next, a GFP-heteroduplex repair assay and microsatellite stability test were used to measure the MMR activities in cells. To investigate the mechanisms, western blotting, the GFP-heteroduplex repair assay, and chromatin immunoprecipitation were used.
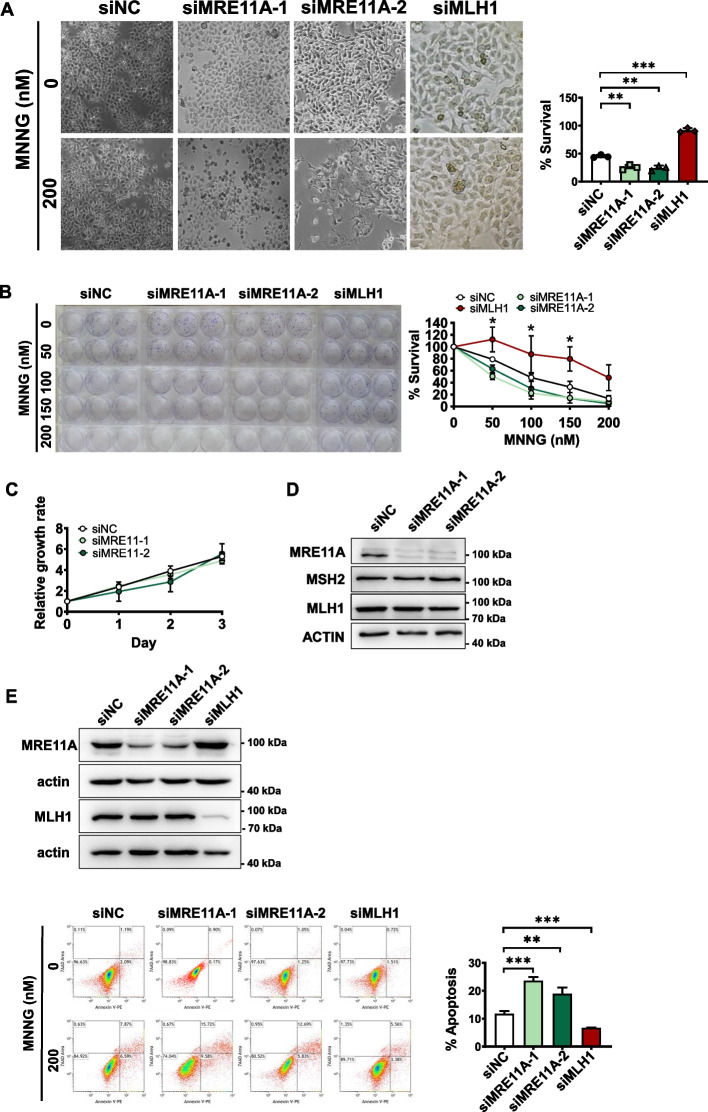
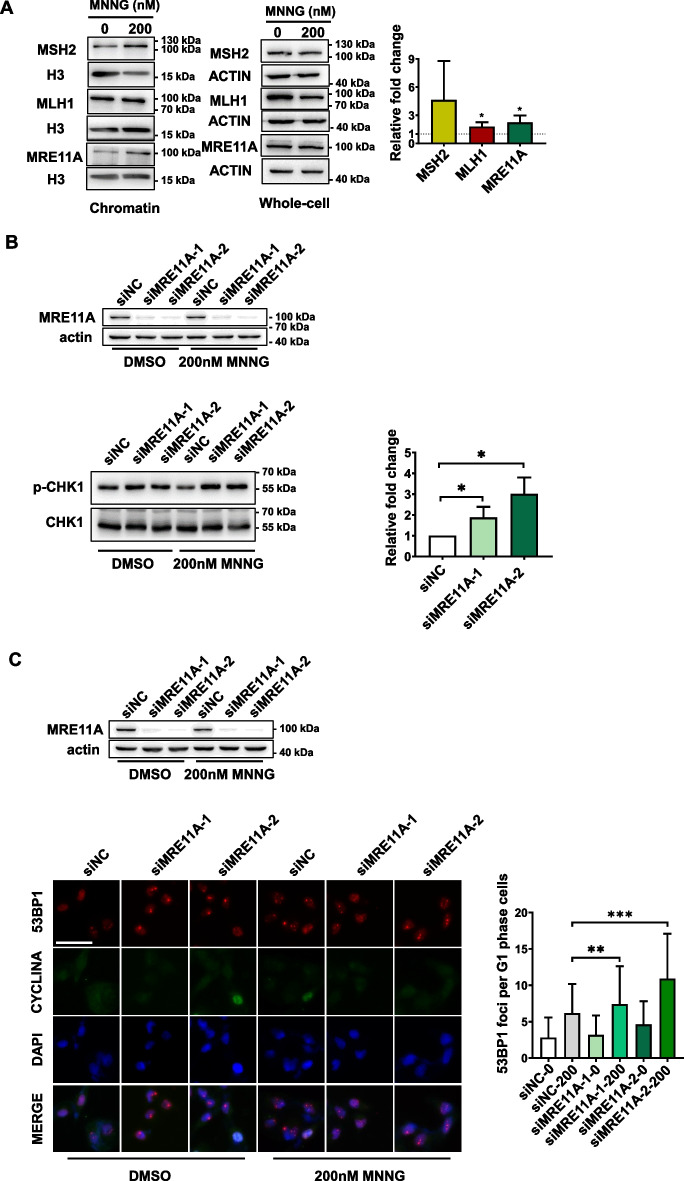
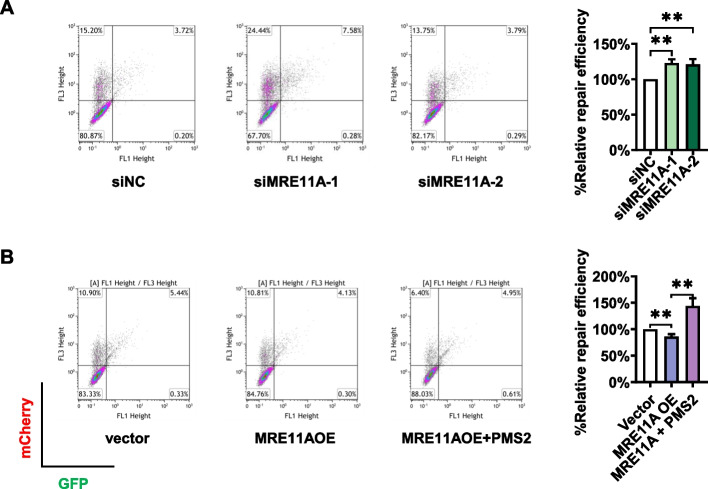
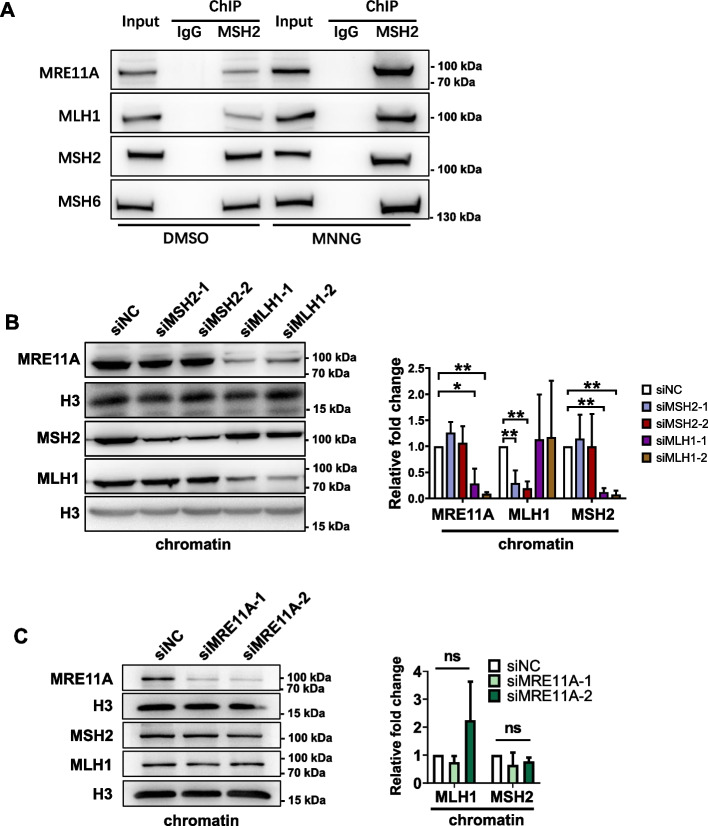
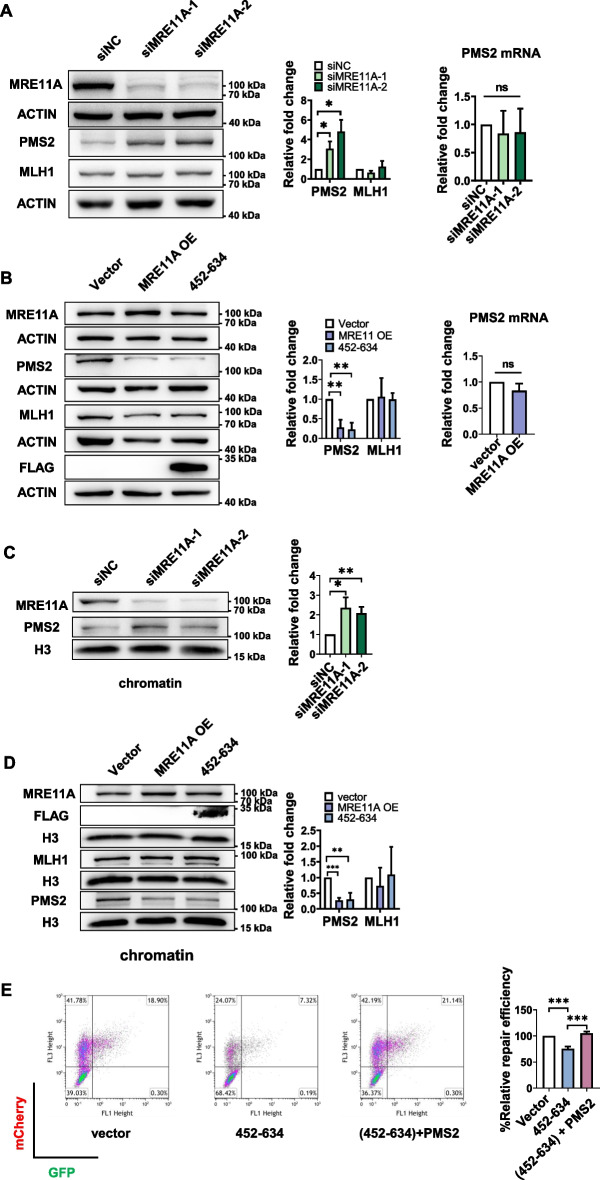
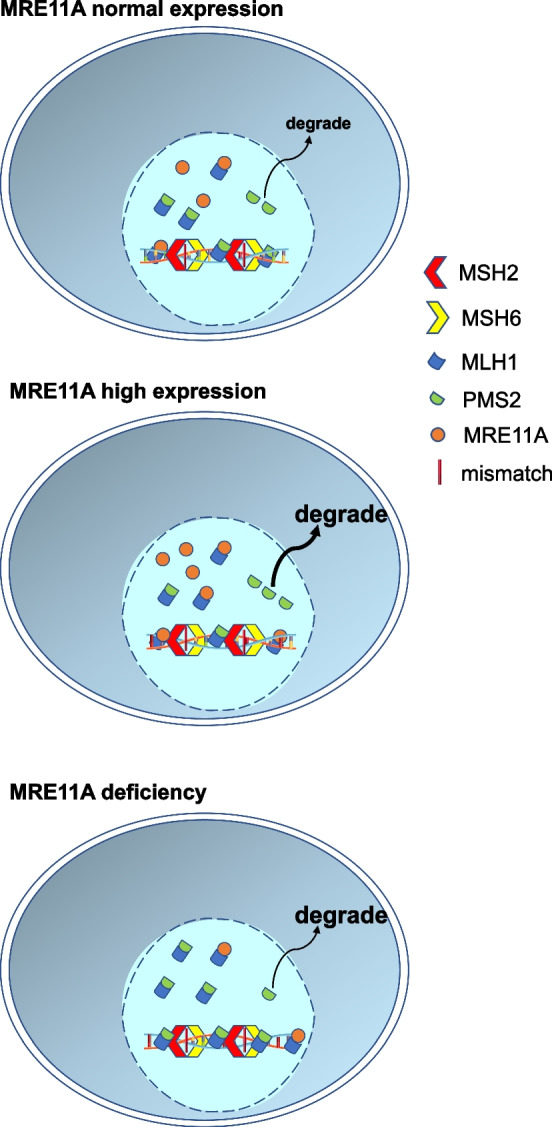
Results: We show that knockdown of MRE11A increased the sensitivity of HeLa cells to MNNG treatment, as well as the MNNG-induced DNA damage and apoptosis, implying a potential role of MRE11 in MMR. Moreover, we found that MRE11A was largely recruited to chromatin and negatively regulated the DNA damage signals within the first cell cycle after MNNG treatment. We also showed that knockdown of MRE11A increased, while overexpressing MRE11A decreased, MMR activity in HeLa cells, suggesting that MRE11A negatively regulates MMR activity. Furthermore, we show that recruitment of MRE11A to chromatin requires MLH1 and that MRE11A competes with PMS2 for binding to MLH1. This decreases PMS2 levels in whole cells and on chromatin, and consequently comprises MMR activity.
Conclusions: Our findings reveal that MRE11A is a negative regulator of human MMR.
Keywords: Alkylating agents; DNA mismatch repair; DNA repair; MRE11A; PMS2.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
Rapid induction of chromatin-associated DNA mismatch repair proteins after MNNG treatment.DNA Repair (Amst). 2008 Jun 1;7(6):951-69. doi: 10.1016/j.dnarep.2008.03.023. Epub 2008 May 12. DNA Repair (Amst). 2008. PMID: 18468964 Free PMC article.
-
N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) triggers MSH2 and Cdt2 protein-dependent degradation of the cell cycle and mismatch repair (MMR) inhibitor protein p21Waf1/Cip1.J Biol Chem. 2011 Aug 26;286(34):29531-9. doi: 10.1074/jbc.M111.221341. Epub 2011 Jul 2. J Biol Chem. 2011. PMID: 21725088 Free PMC article.
-
MMR/c-Abl-dependent activation of ING2/p73alpha signaling regulates the cell death response to N-methyl-N'-nitro-N-nitrosoguanidine.Exp Cell Res. 2009 Nov 1;315(18):3163-75. doi: 10.1016/j.yexcr.2009.09.010. Epub 2009 Sep 17. Exp Cell Res. 2009. PMID: 19766113 Free PMC article.
-
Mismatch repair pathway: molecules, functions, and role in colorectal carcinogenesis.Eur J Cancer Prev. 2014 Jul;23(4):246-57. doi: 10.1097/CEJ.0000000000000019. Eur J Cancer Prev. 2014. PMID: 24614649 Review.
-
Targeting Mismatch Repair defects: A novel strategy for personalized cancer treatment.DNA Repair (Amst). 2016 Feb;38:135-139. doi: 10.1016/j.dnarep.2015.11.026. Epub 2015 Dec 2. DNA Repair (Amst). 2016. PMID: 26698647 Review.
Cited by
-
Microsatellite Instability in the Tumor Microenvironment: The Role of Inflammation and the Microbiome.Cancer Med. 2025 Apr;14(8):e70603. doi: 10.1002/cam4.70603. Cancer Med. 2025. PMID: 40231893 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
