

Disassembly of the TRIM56-ATR complex promotes cytoDNA/cGAS/STING axis-dependent intervertebral disc inflammatory degeneration
- PMID: 38488012
- PMCID: PMC10940101
- DOI: 10.1172/JCI165140
Disassembly of the TRIM56-ATR complex promotes cytoDNA/cGAS/STING axis-dependent intervertebral disc inflammatory degeneration
Abstract
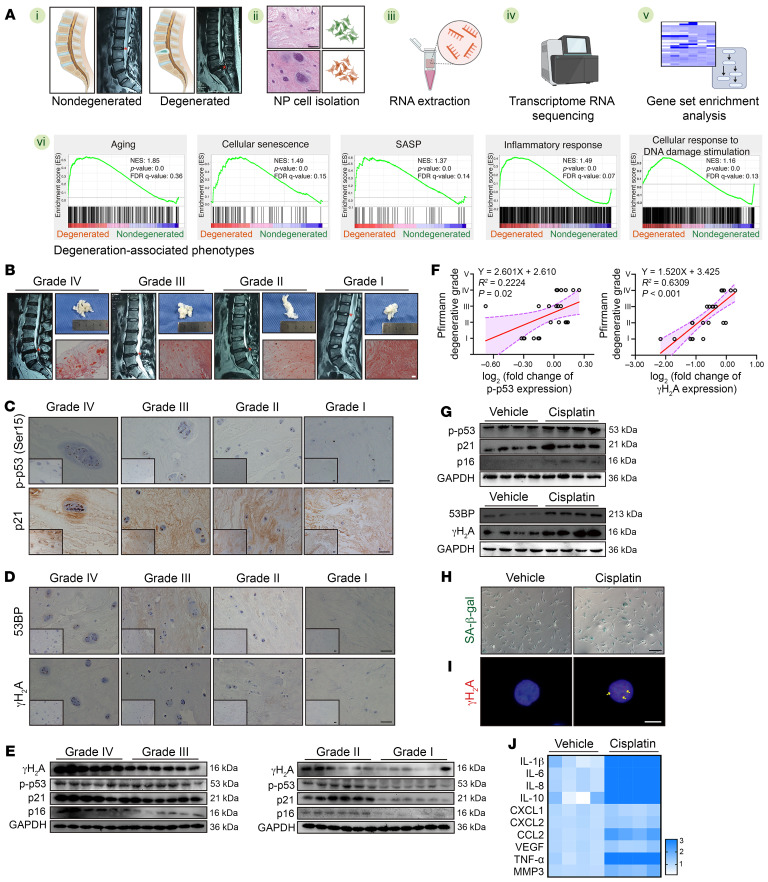
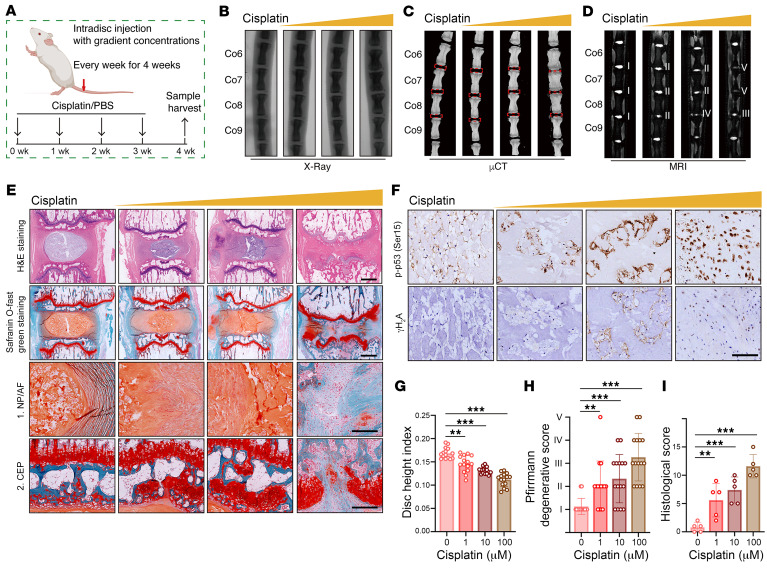
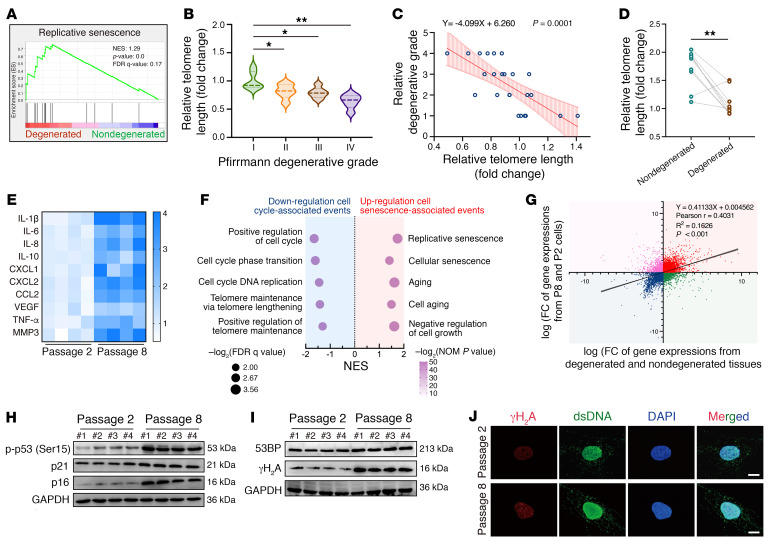
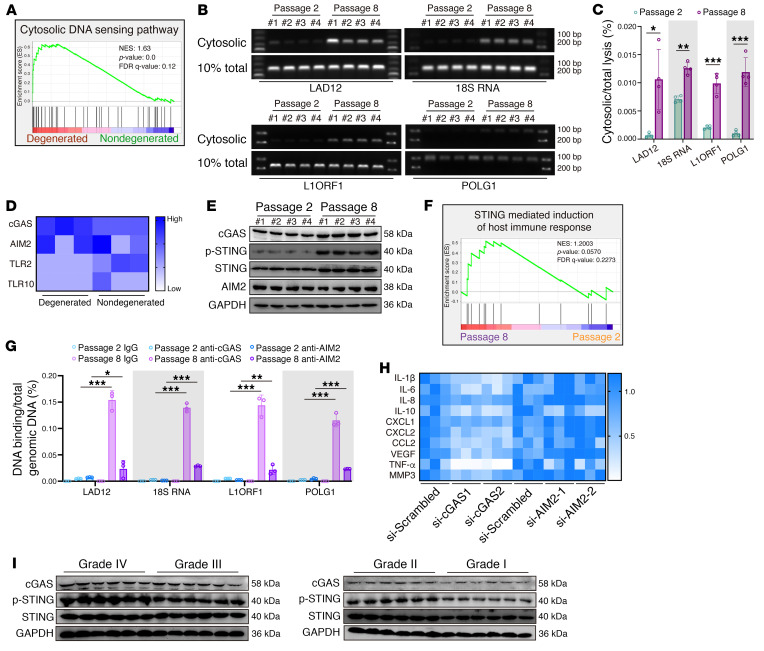
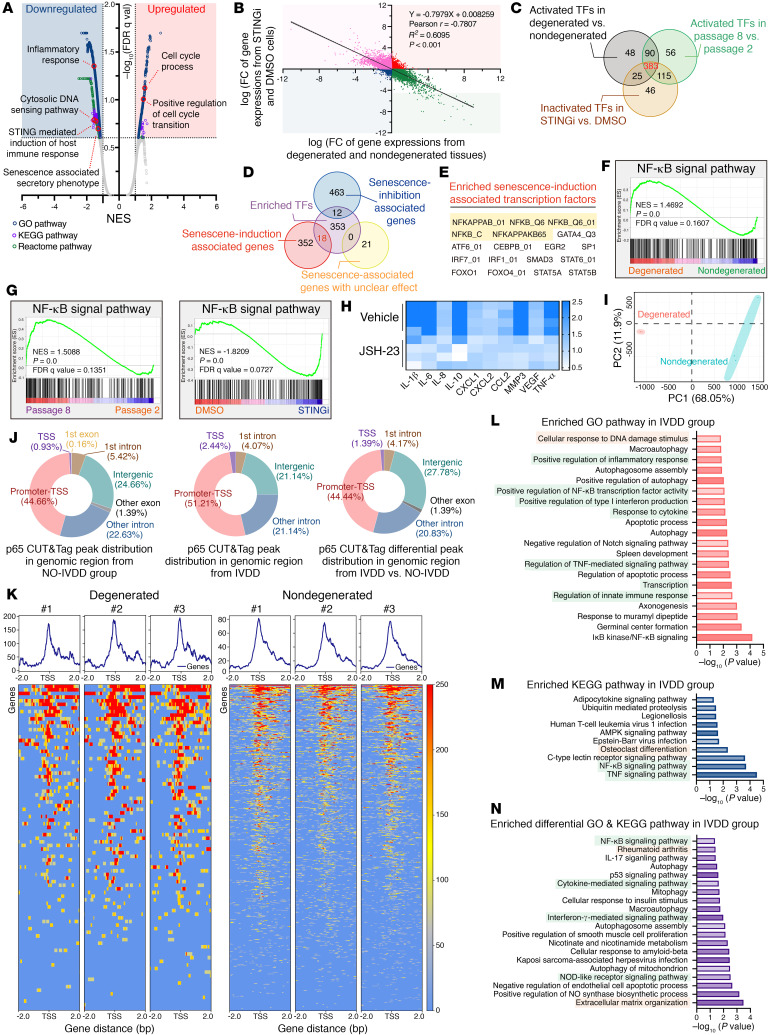
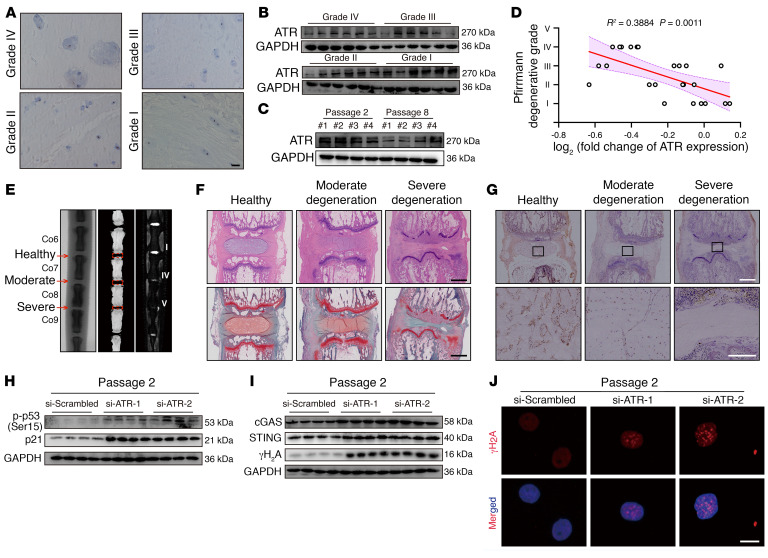
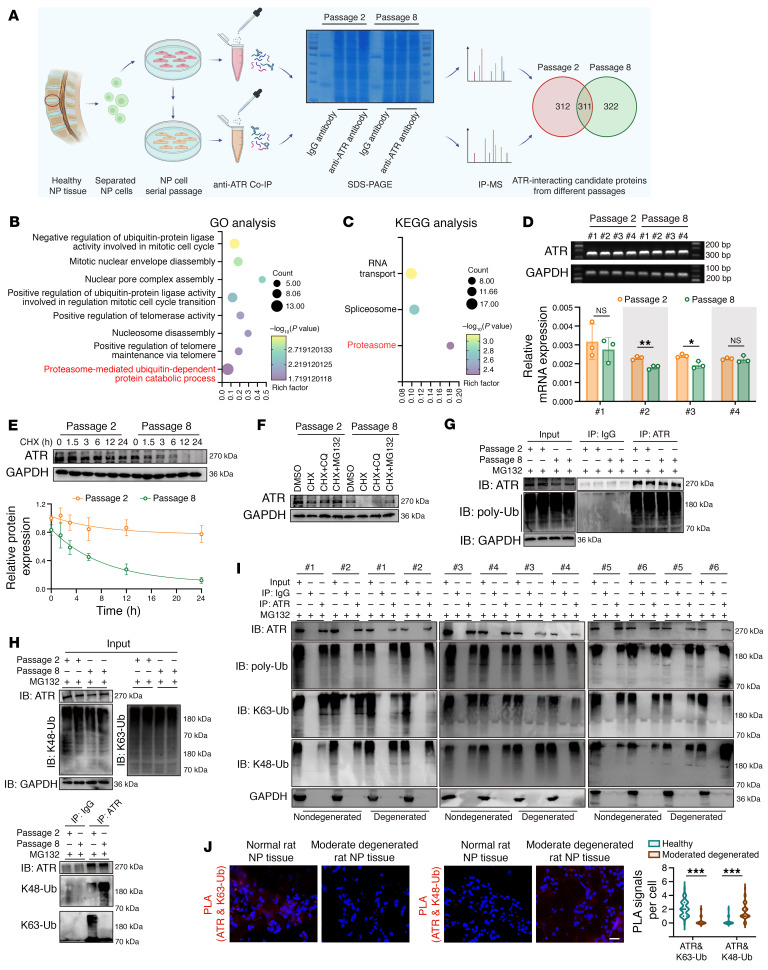
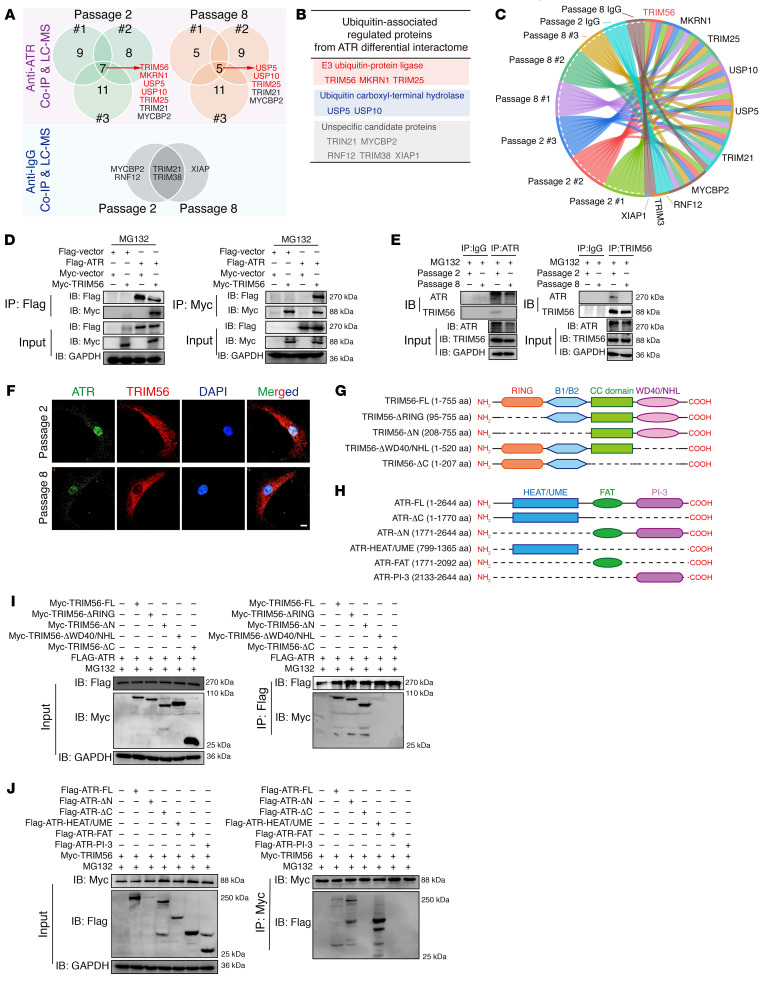
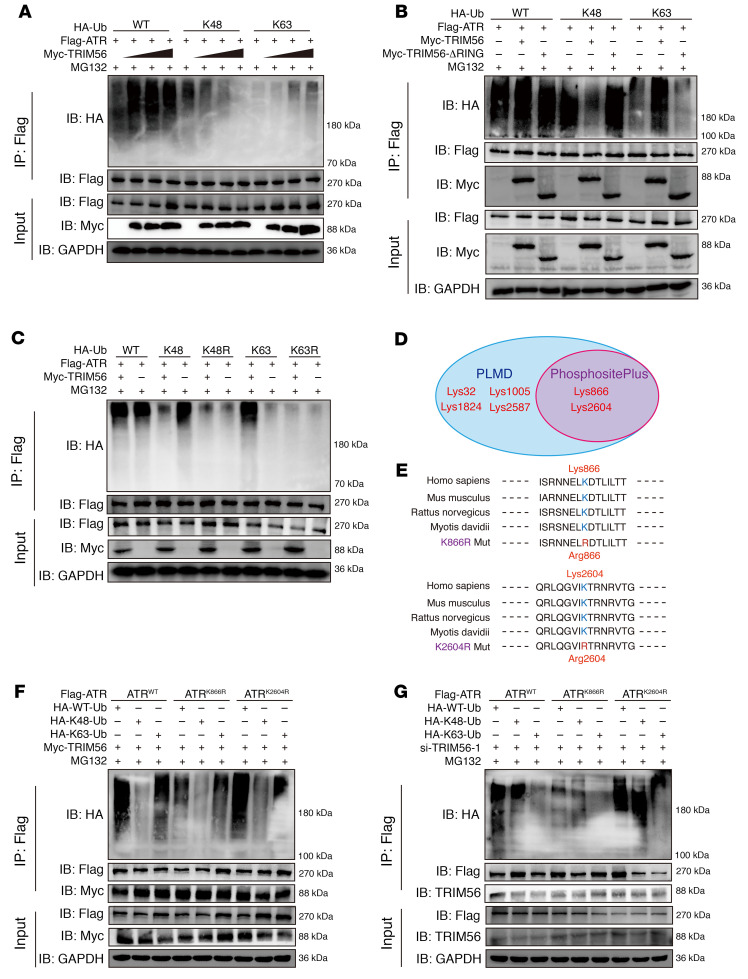
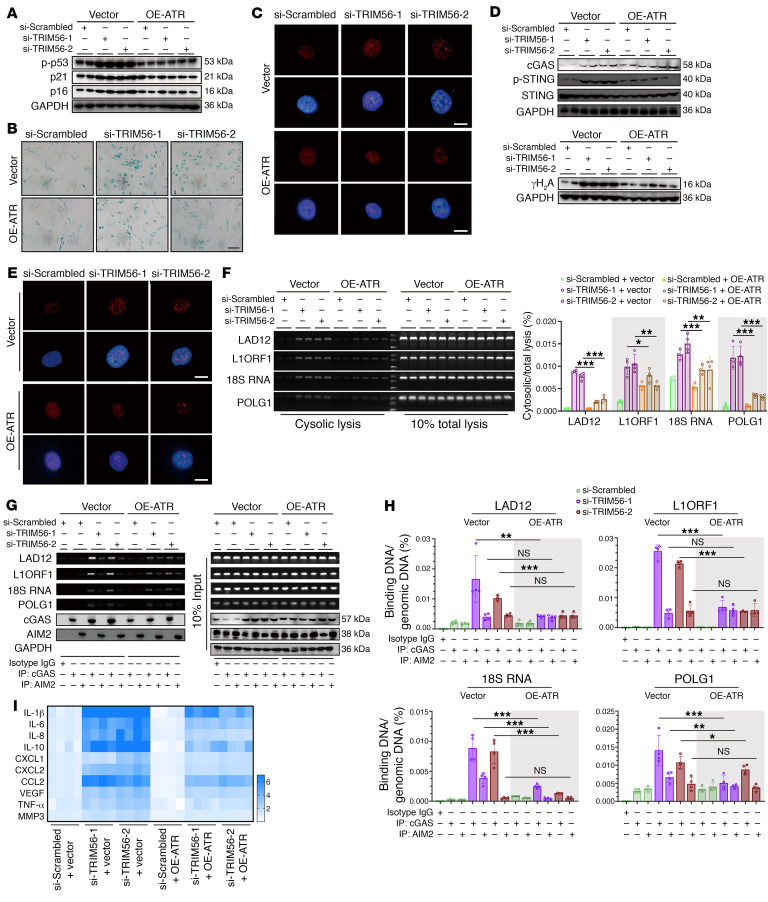
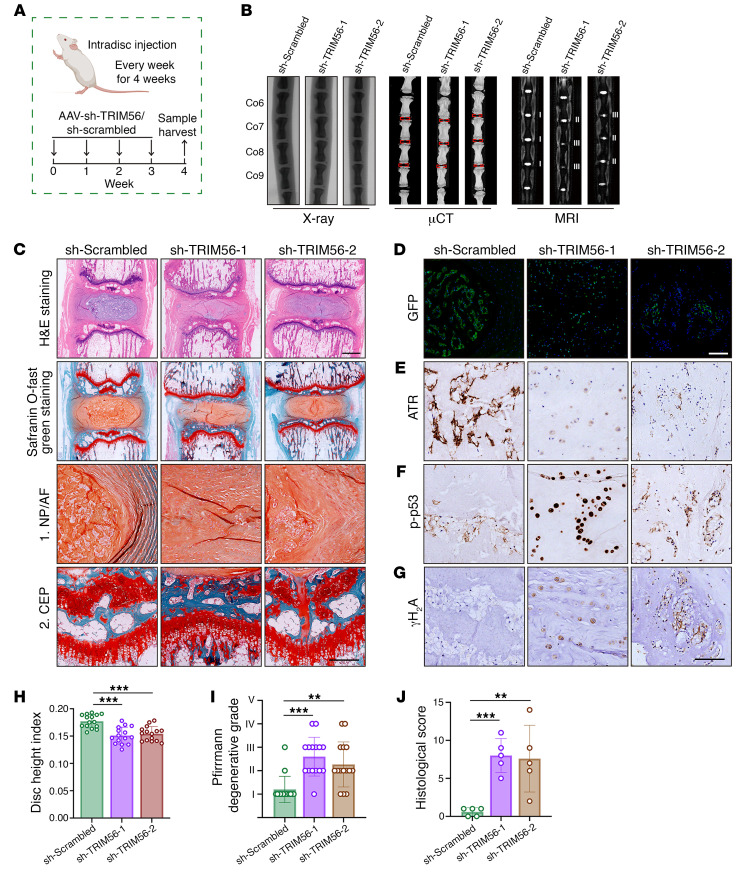
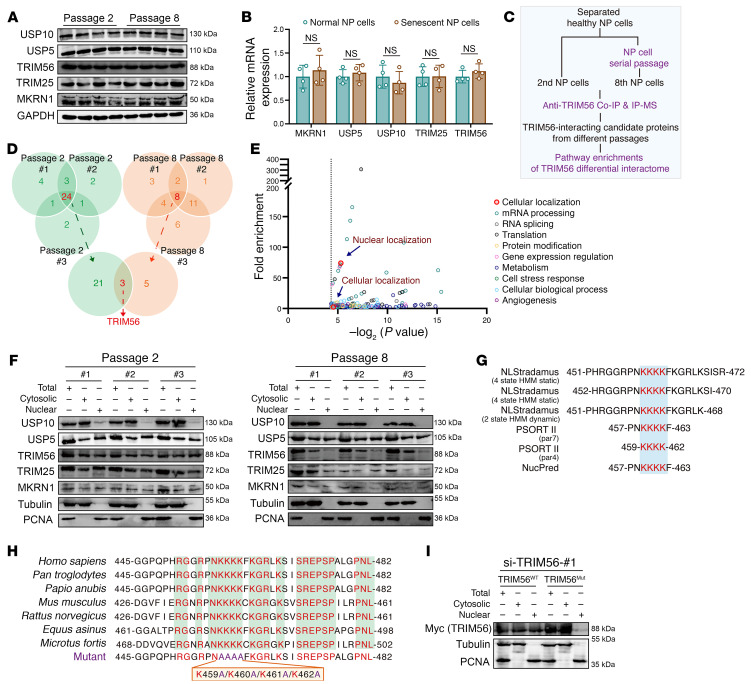
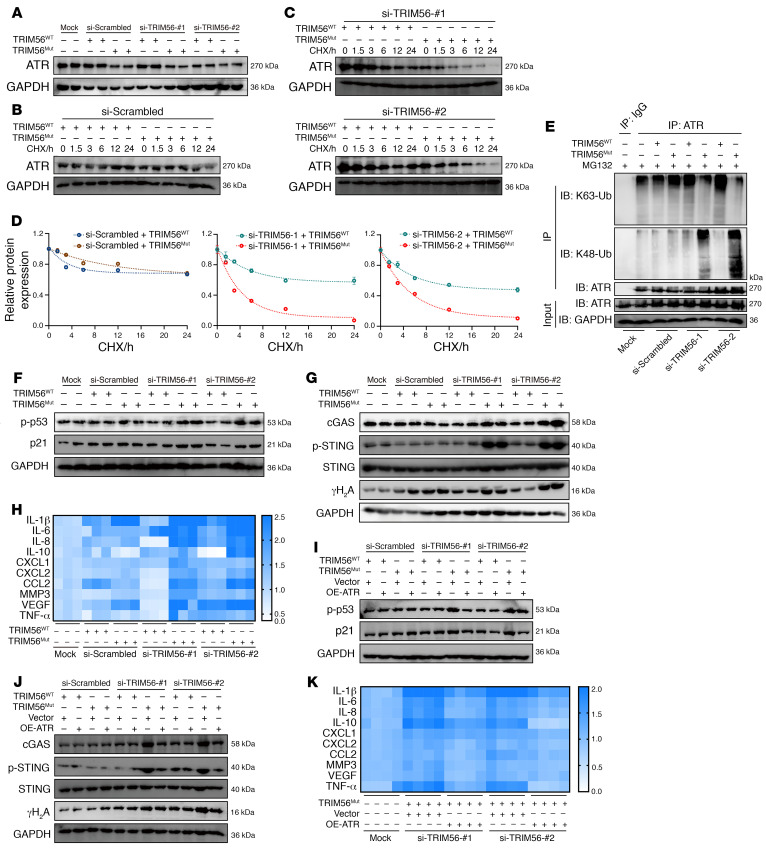
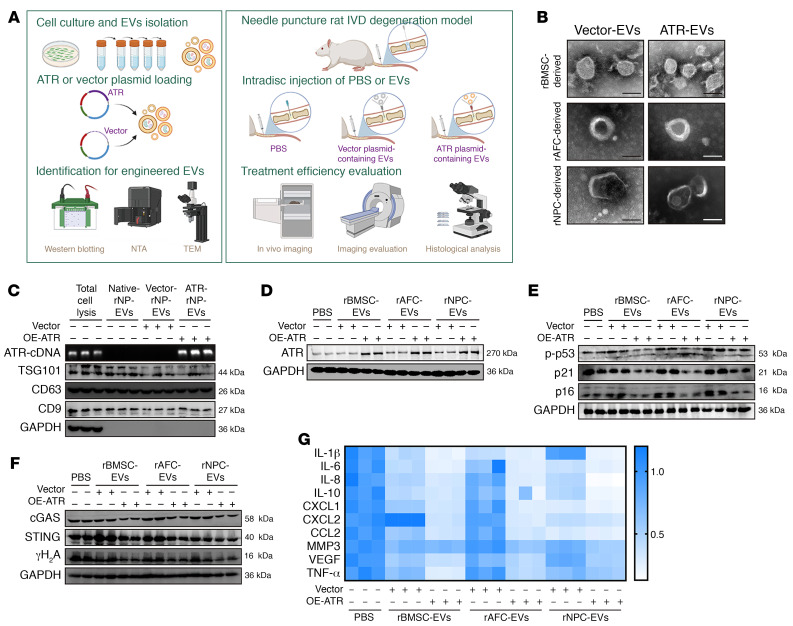
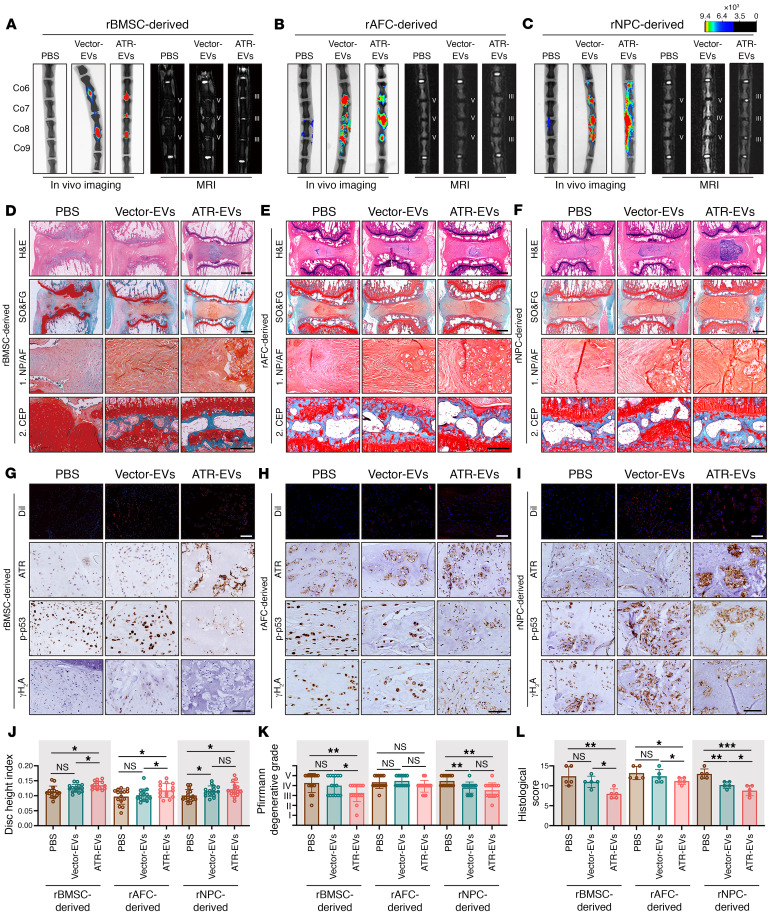
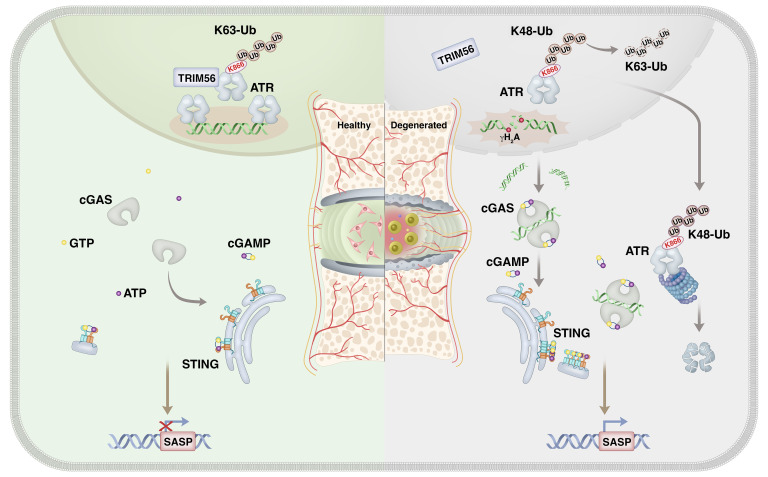
As the leading cause of disability worldwide, low back pain (LBP) is recognized as a pivotal socioeconomic challenge to the aging population and is largely attributed to intervertebral disc degeneration (IVDD). Elastic nucleus pulposus (NP) tissue is essential for the maintenance of IVD structural and functional integrity. The accumulation of senescent NP cells with an inflammatory hypersecretory phenotype due to aging and other damaging factors is a distinctive hallmark of IVDD initiation and progression. In this study, we reveal a mechanism of IVDD progression in which aberrant genomic DNA damage promoted NP cell inflammatory senescence via activation of the cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING) axis but not of absent in melanoma 2 (AIM2) inflammasome assembly. Ataxia-telangiectasia-mutated and Rad3-related protein (ATR) deficiency destroyed genomic integrity and led to cytosolic mislocalization of genomic DNA, which acted as a powerful driver of cGAS/STING axis-dependent inflammatory phenotype acquisition during NP cell senescence. Mechanistically, disassembly of the ATR-tripartite motif-containing 56 (ATR-TRIM56) complex with the enzymatic liberation of ubiquitin-specific peptidase 5 (USP5) and TRIM25 drove changes in ATR ubiquitination, with ATR switching from K63- to K48-linked modification, c thereby promoting ubiquitin-proteasome-dependent dynamic instability of ATR protein during NP cell senescence progression. Importantly, an engineered extracellular vesicle-based strategy for delivering ATR-overexpressing plasmid cargo efficiently diminished DNA damage-associated NP cell senescence and substantially mitigated IVDD progression, indicating promising targets and effective approaches to ameliorate the chronic pain and disabling effects of IVDD.
Keywords: Bone biology; Bone disease; Orthopedics.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
