

Annotation of 2,507 Saccharomyces cerevisiae genomes
- PMID: 38488392
- PMCID: PMC10986567
- DOI: 10.1128/spectrum.03582-23
Annotation of 2,507 Saccharomyces cerevisiae genomes
Erratum in
-
Erratum for Wang et al., "Annotation of 2,507 Saccharomyces cerevisiae genomes".Microbiol Spectr. 2024 Nov 12;12(12):e0237424. doi: 10.1128/spectrum.02374-24. Online ahead of print. Microbiol Spectr. 2024. PMID: 39527776 Free PMC article. No abstract available.
Abstract
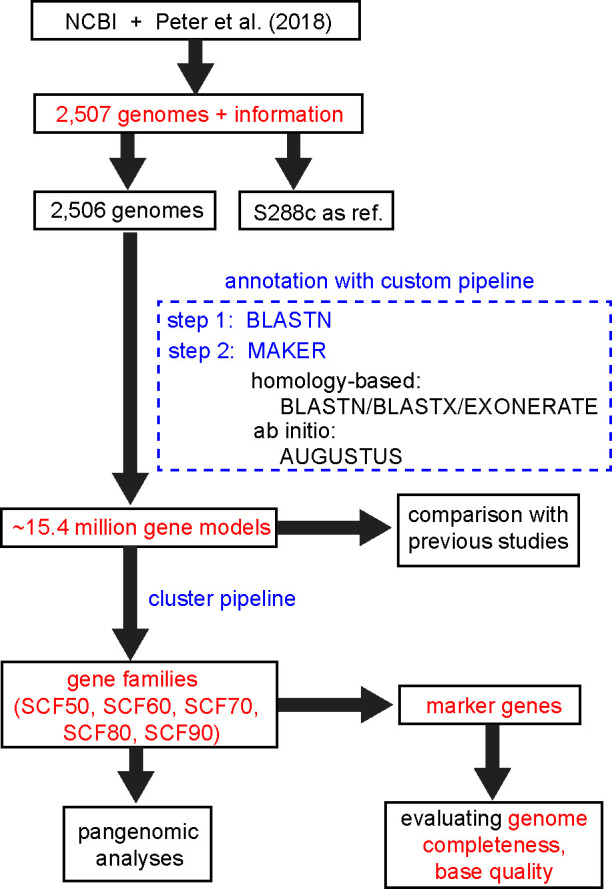
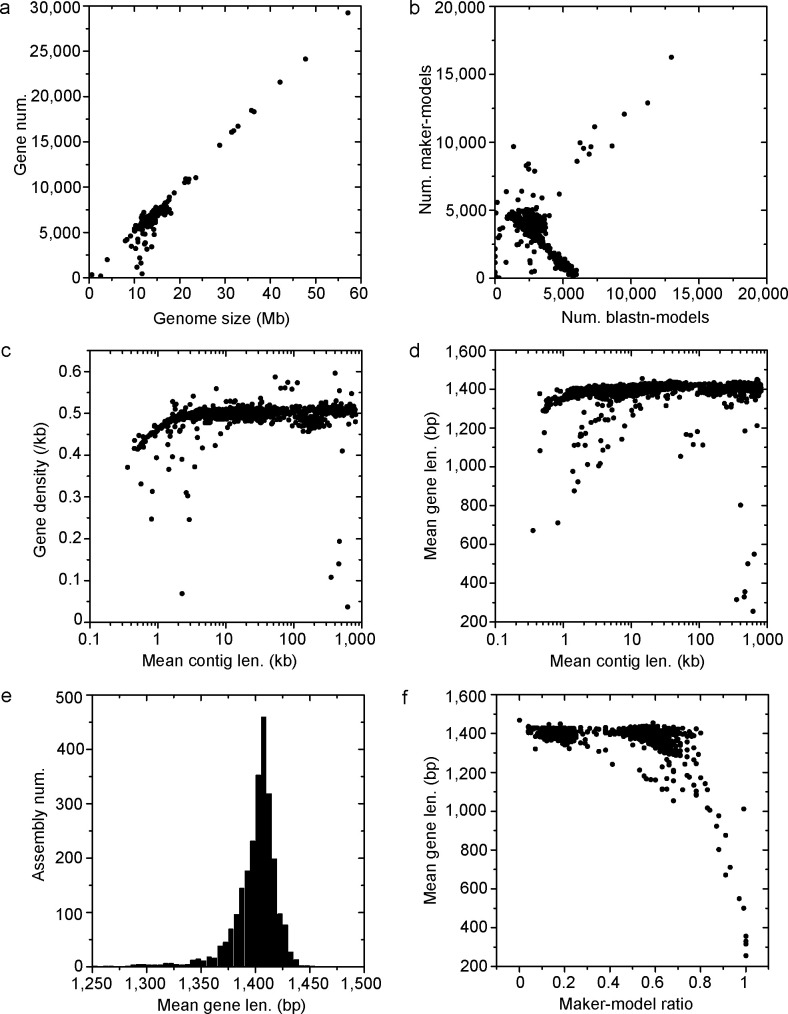
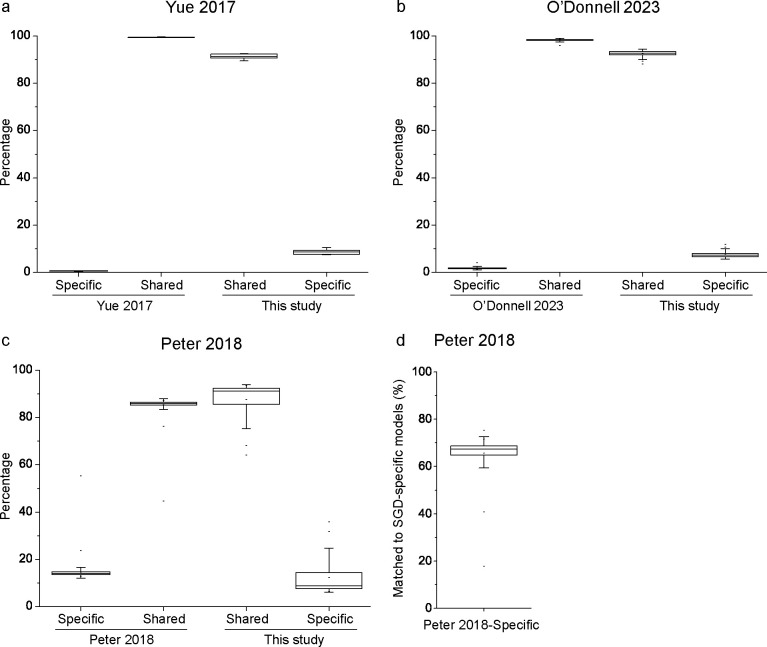
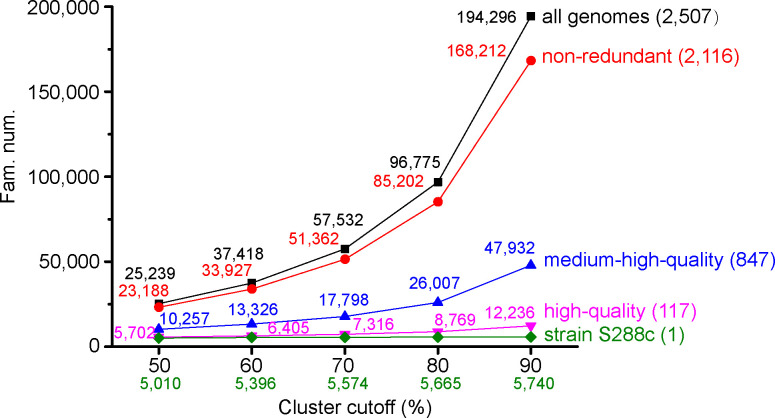
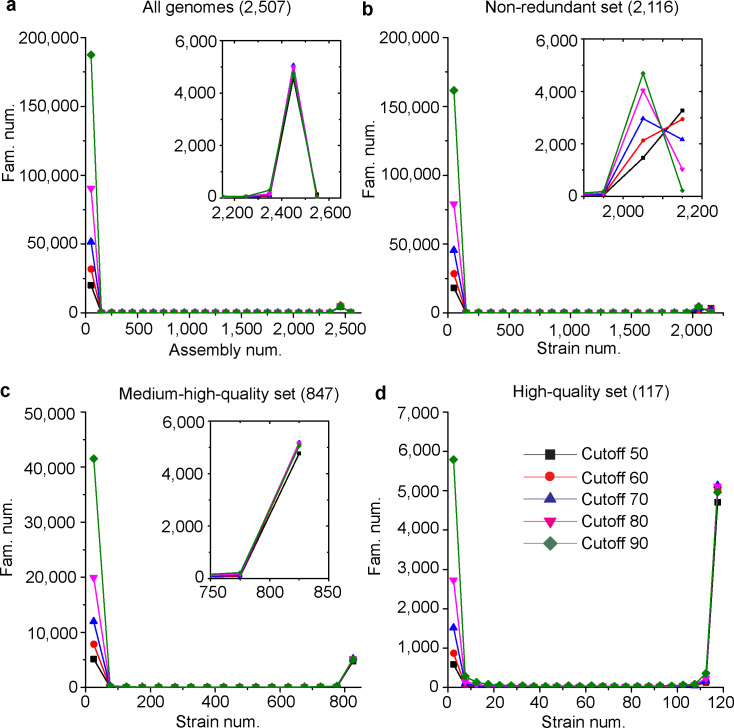
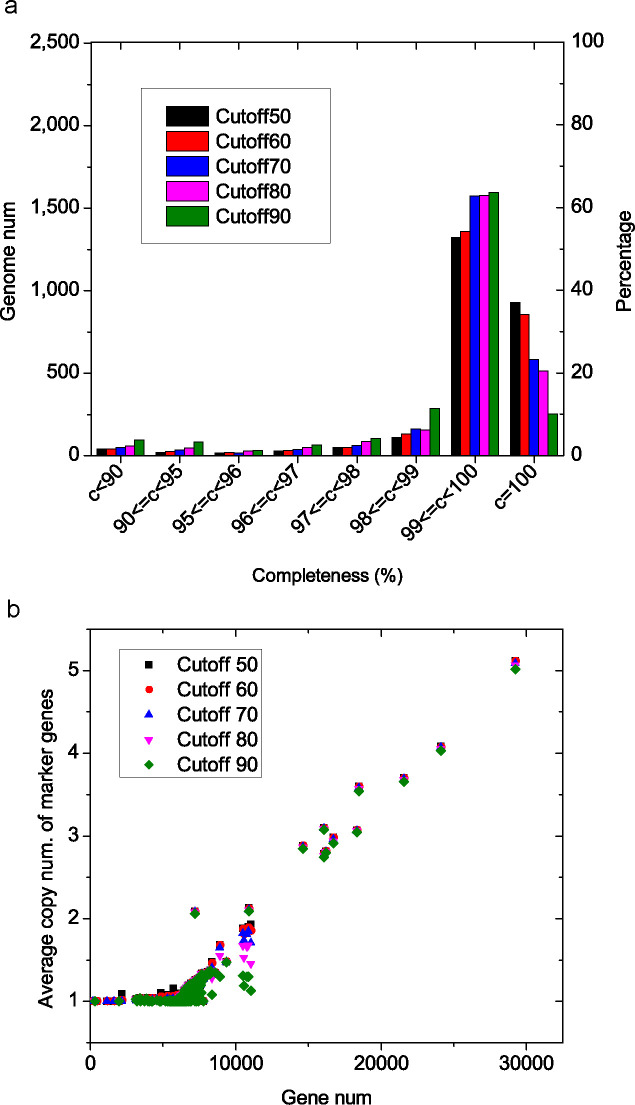
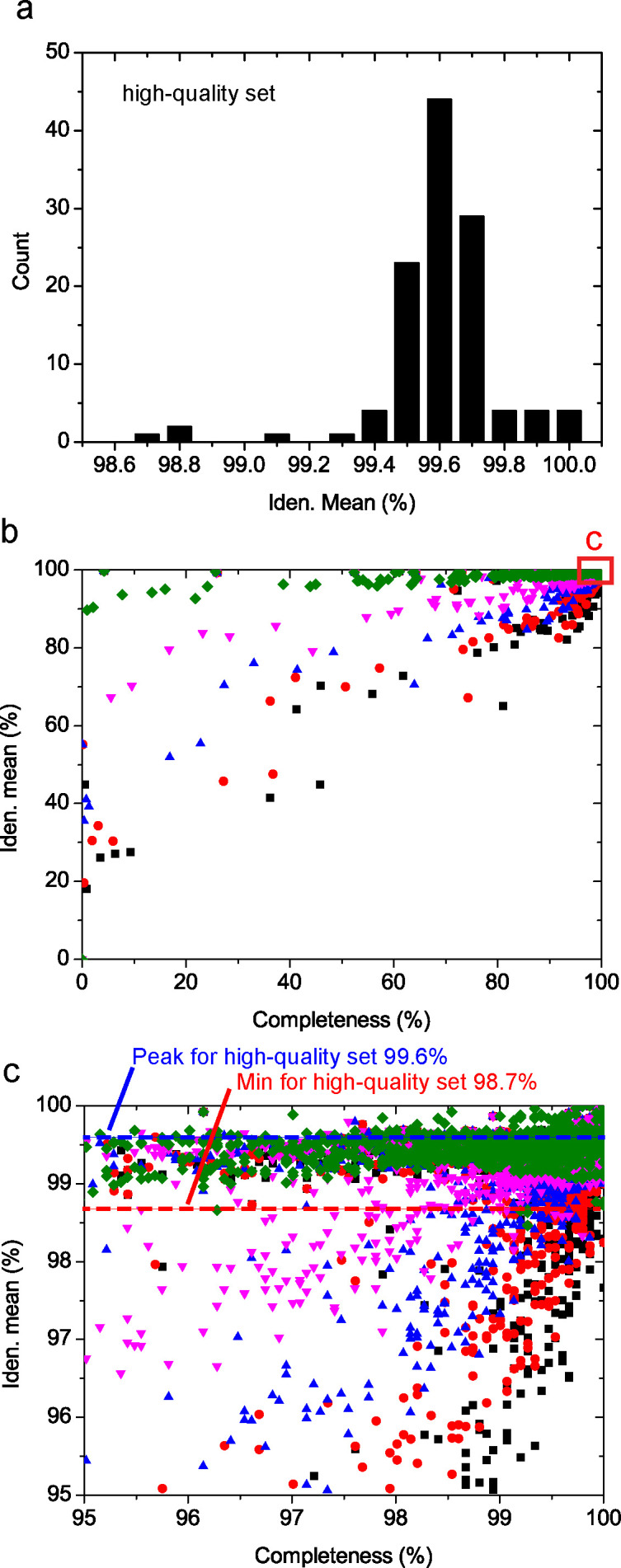
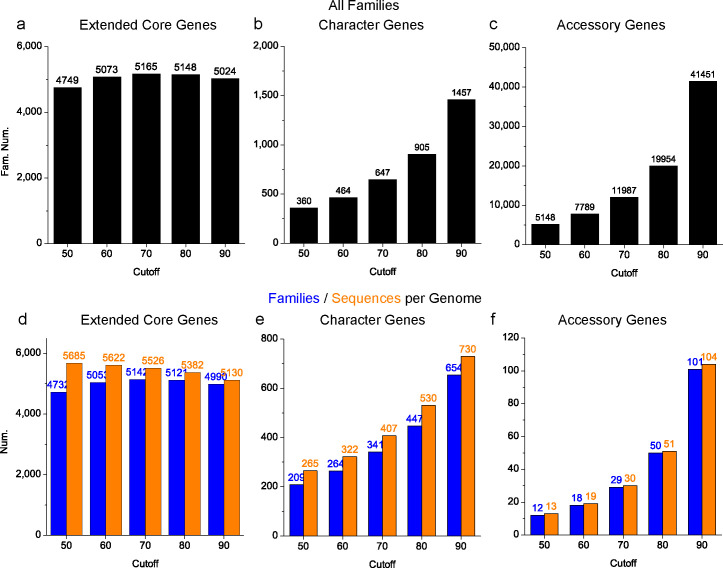
Saccharomyces cerevisiae (baker's yeast, budding yeast) is one of the most important model organisms for biological research and is a crucial microorganism in industry. Currently, a huge number of Saccharomyces cerevisiae genome sequences are available at the public domain. However, these genomes are distributed at different websites and a large number of them are released without annotation information. To provide one complete annotated genome data resource, we collected 2,507 Saccharomyces cerevisiae genome assemblies and re-annotated 2,506 assemblies using a custom annotation pipeline, producing a total of 15,407,164 protein-coding gene models. With a custom pipeline, all these gene sequences were clustered into families. A total of 1,506 single-copy genes were selected as marker genes, which were then used to evaluate the genome completeness and base qualities of all assemblies. Pangenomic analyses were performed based on a selected subset of 847 medium-high-quality genomes. Statistical comparisons revealed a number of gene families showing copy number variations among different organism sources. To the authors' knowledge, this study represents the largest genome annotation project of S. cerevisiae so far, providing rich genomic resources for the future studies of the model organism S. cerevisiae and its relatives.IMPORTANCESaccharomyces cerevisiae (baker's yeast, budding yeast) is one of the most important model organisms for biological research and is a crucial microorganism in industry. Though a huge number of Saccharomyces cerevisiae genome sequences are available at the public domain, these genomes are distributed at different websites and most are released without annotation, hindering the efficient reuse of these genome resources. Here, we collected 2,507 genomes for Saccharomyces cerevisiae, performed genome annotation, and evaluated the genome qualities. All the obtained data have been deposited at public repositories and are freely accessible to the community. This study represents the largest genome annotation project of S. cerevisiae so far, providing one complete annotated genome data set for S. cerevisiae, an important workhorse for fundamental biology, biotechnology, and industry.
Keywords: Saccharomyces cerevisiae; annotation; genome.
Conflict of interest statement
Xiaoping Hou, Yang He, Jun-Hong Yu, Shumin Hu, and Hua Yin are employed by Tsingtao Brewery Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, Cruaud C, Labadie K, Aury J-M, Istace B, Lebrigand K, Barbry P, Engelen S, Lemainque A, Wincker P, Liti G, Schacherer J. 2018. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556:339–344. doi:10.1038/s41586-018-0030-5 - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
