

Tau induces inflammasome activation and microgliosis through acetylating NLRP3
- PMID: 38488468
- PMCID: PMC10941548
- DOI: 10.1002/ctm2.1623
Tau induces inflammasome activation and microgliosis through acetylating NLRP3
Abstract
Background: Alzheimer's disease (AD) and related Tauopathies are characterised by the pathologically hyperphosphorylated and aggregated microtubule-associated protein Tau, which is accompanied by neuroinflammation mediated by activated microglia. However, the role of Tau pathology in microglia activation or their causal relationship remains largely elusive.
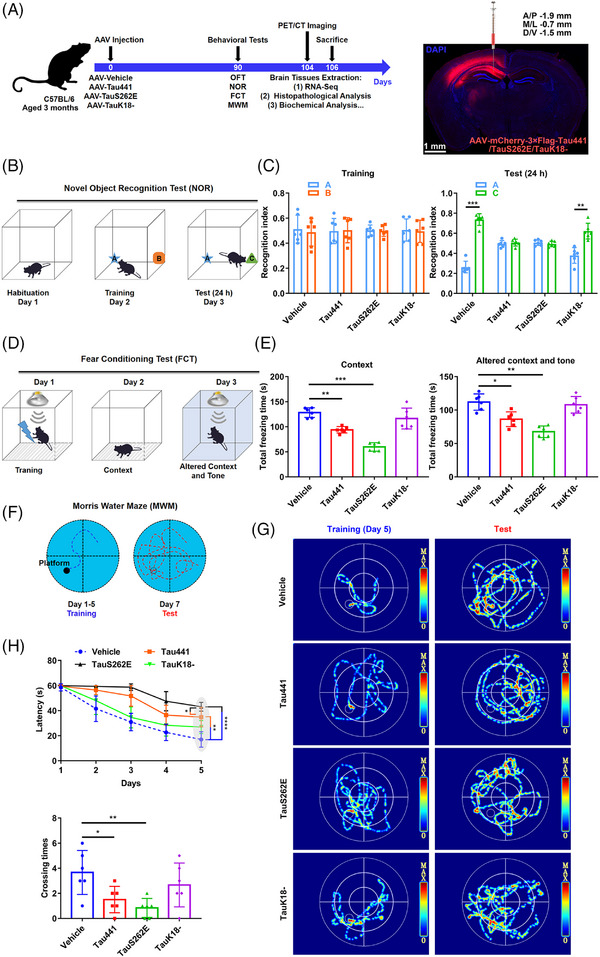
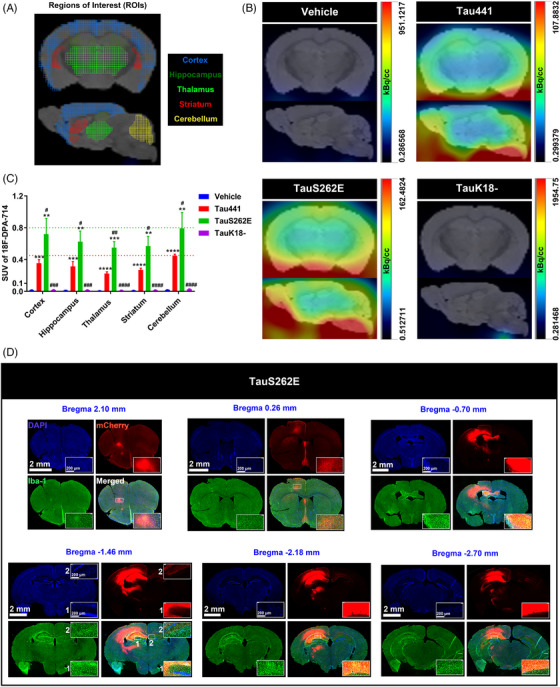
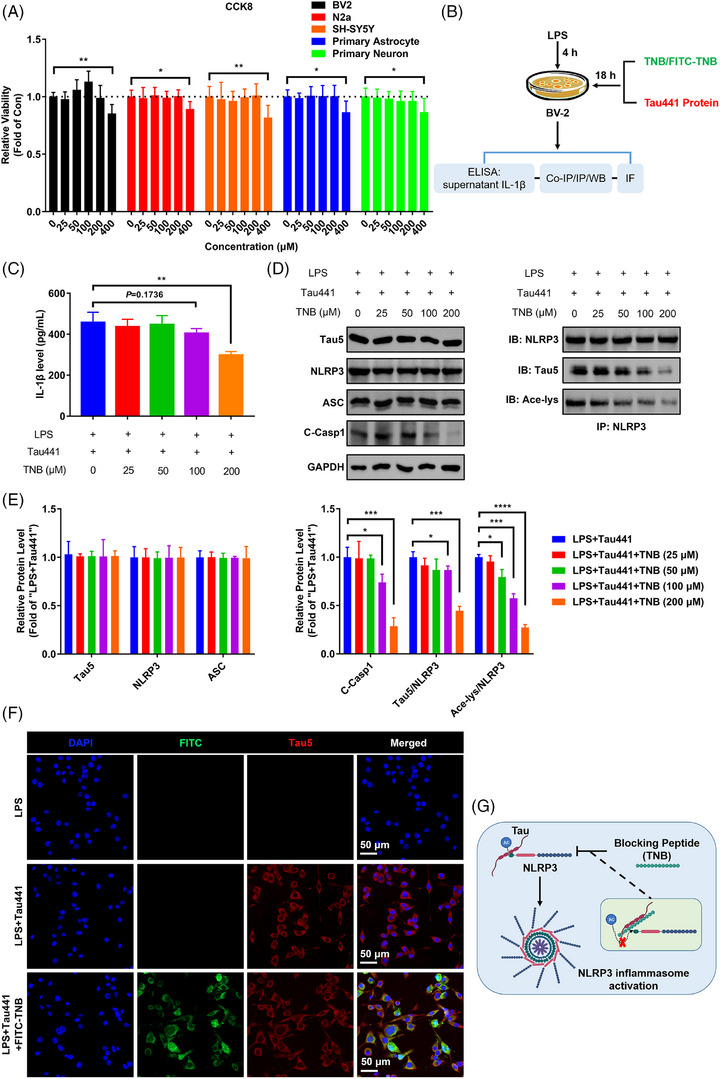
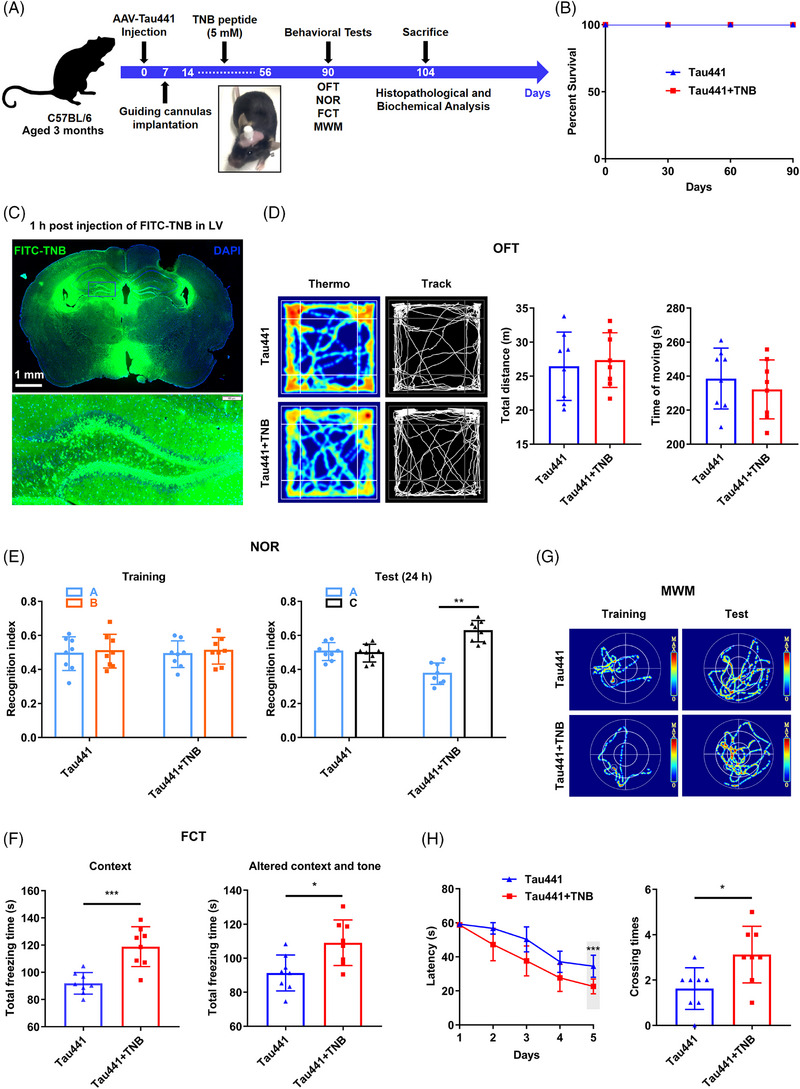
Methods: The levels of nucleotide-binding oligomerisation domain (NOD)-like receptor pyrin domain containing 3 (NLRP3) acetylation and inflammasome activation in multiple cell models with Tau proteins treatment, transgenic mice with Tauopathy, and AD patients were measured by Western blotting and enzyme-linked immunosorbent assay. In addition, the acetyltransferase activity of Tau and NLRP3 acetylation sites were confirmed using the test-tube acetylation assay, co-immunoprecipitation, immunofluorescence (IF) staining, mass spectrometry and molecular docking. The Tau-overexpressing mouse model was established by overexpression of human Tau proteins in mouse hippocampal CA1 neurons through the adeno-associated virus injection. The cognitive functions of Tau-overexpressing mice were assessed in various behavioural tests, and microglia activation was analysed by Iba-1 IF staining and [18F]-DPA-714 positron emission tomography/computed tomography imaging. A peptide that blocks the interaction between Tau and NLRP3 was synthesised to determine the in vitro and in vivo effects of Tau-NLRP3 interaction blockade on NLRP3 acetylation, inflammasome activation, microglia activation and cognitive function.
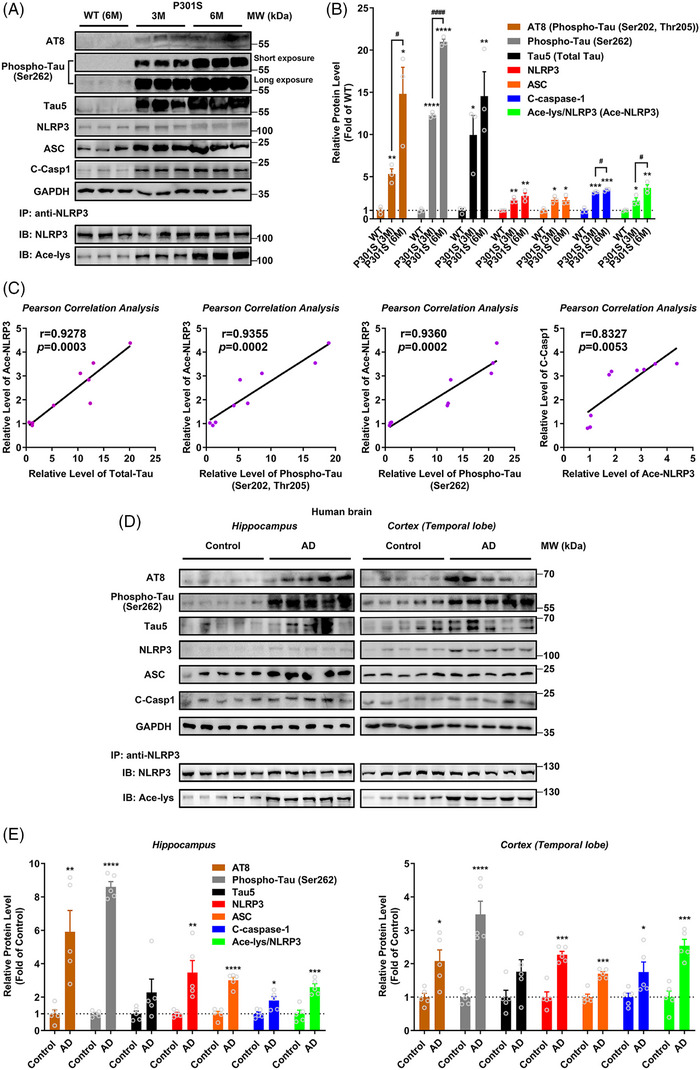
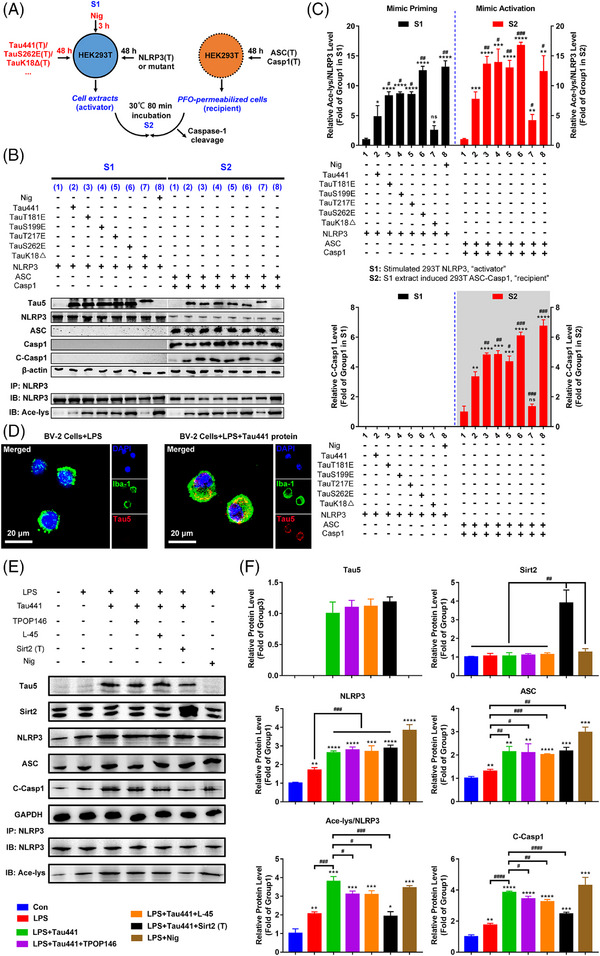
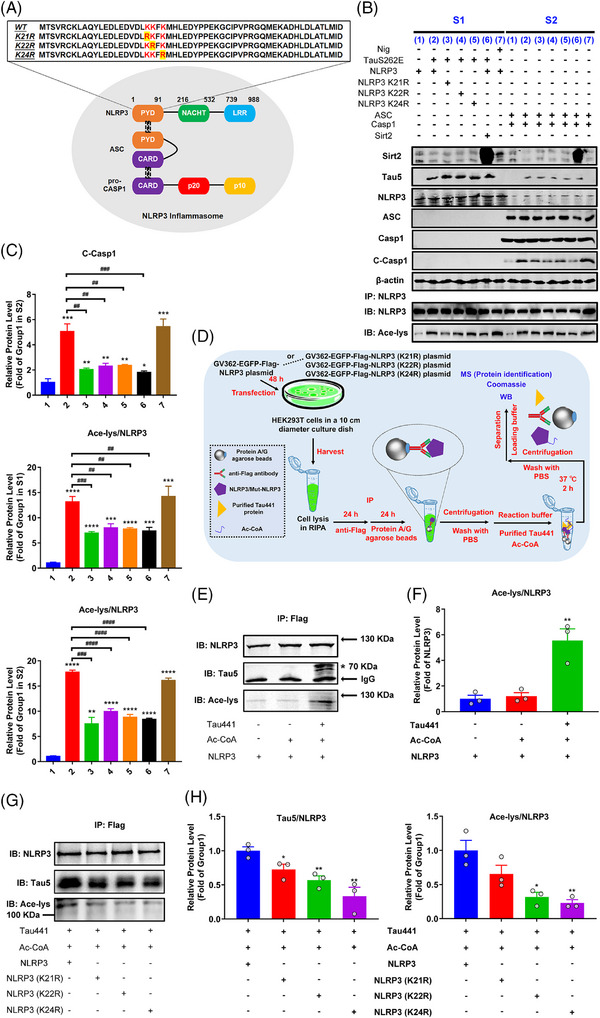
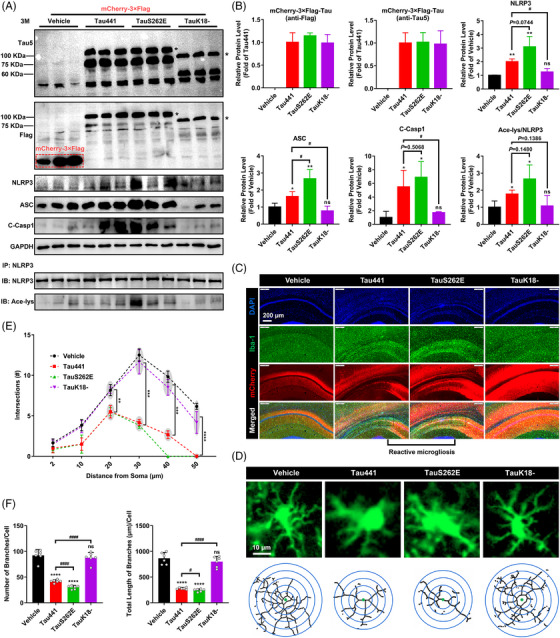
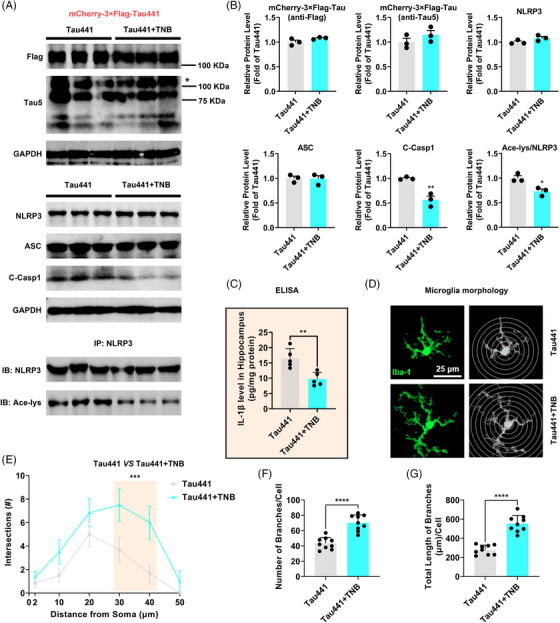
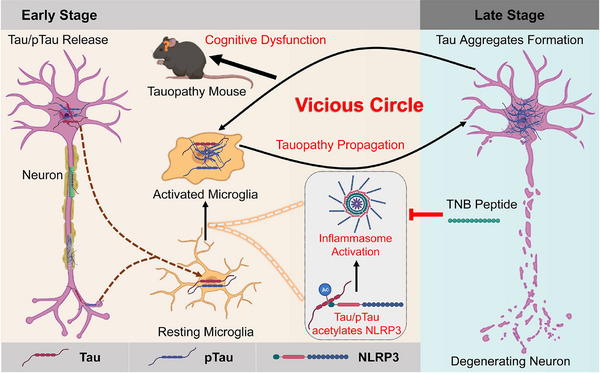
Results: Excessively elevated NLRP3 acetylation and inflammasome activation were observed in 3xTg-AD mice, microtubule-associated protein Tau P301S (PS19) mice and AD patients. It was further confirmed that mimics of 'early' phosphorylated-Tau proteins which increase at the initial stage of diseases with Tauopathy, including TauT181E, TauS199E, TauT217E and TauS262E, significantly promoted Tau-K18 domain acetyltransferase activity-dependent NLRP3 acetylation and inflammasome activation in HEK293T and BV-2 microglial cells. In addition, Tau protein could directly acetylate NLRP3 at the K21, K22 and K24 sites at its PYD domain and thereby induce inflammasome activation in vitro. Overexpression of human Tau proteins in mouse hippocampal CA1 neurons resulted in impaired cognitive function, Tau transmission to microglia and microgliosis with NLRP3 acetylation and inflammasome activation. As a targeted intervention, competitive binding of a designed Tau-NLRP3-binding blocking (TNB) peptide to block the interaction of Tau protein with NLRP3 inhibited the NLRP3 acetylation and downstream inflammasome activation in microglia, thereby alleviating microglia activation and cognitive impairment in mice.
Conclusions: In conclusion, our findings provide evidence for a novel role of Tau in the regulation of microglia activation through acetylating NLRP3, which has potential implications for early intervention and personalised treatment of AD and related Tauopathies.
Keywords: Alzheimer's disease; NLRP3 inflammasome; Tauopathies; acetylation; microglia.
© 2024 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare they have no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- 82171426/National Natural Science Foundation of China
- 31970964/National Natural Science Foundation of China
- 61860206009/National Natural Science Foundation of China
- JCYJ20220530160805012/Science, Technology and Innovation Commission of Shenzhen Municipality
- 2023020201010196/Basic Research Programme Funded by Wuhan Science and Technology Bureau
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous