

Casein kinase 2 phosphorylates and induces the SALL2 tumor suppressor degradation in colon cancer cells
- PMID: 38493149
- PMCID: PMC10944491
- DOI: 10.1038/s41419-024-06591-z
Casein kinase 2 phosphorylates and induces the SALL2 tumor suppressor degradation in colon cancer cells
Abstract
Spalt-like proteins are Zinc finger transcription factors from Caenorhabditis elegans to vertebrates, with critical roles in development. In vertebrates, four paralogues have been identified (SALL1-4), and SALL2 is the family's most dissimilar member. SALL2 is required during brain and eye development. It is downregulated in cancer and acts as a tumor suppressor, promoting cell cycle arrest and cell death. Despite its critical functions, information about SALL2 regulation is scarce. Public data indicate that SALL2 is ubiquitinated and phosphorylated in several residues along the protein, but the mechanisms, biological consequences, and enzymes responsible for these modifications remain unknown. Bioinformatic analyses identified several putative phosphorylation sites for Casein Kinase II (CK2) located within a highly conserved C-terminal PEST degradation motif of SALL2. CK2 is a serine/threonine kinase that promotes cell proliferation and survival and is often hyperactivated in cancer. We demonstrated that CK2 phosphorylates SALL2 residues S763, T778, S802, and S806 and promotes SALL2 degradation by the proteasome. Accordingly, pharmacological inhibition of CK2 with Silmitasertib (CX-4945) restored endogenous SALL2 protein levels in SALL2-deficient breast MDA-MB-231, lung H1299, and colon SW480 cancer cells. Silmitasertib induced a methuosis-like phenotype and cell death in SW480 cells. However, the phenotype was significantly attenuated in CRISPr/Cas9-mediated SALL2 knockout SW480 cells. Similarly, Sall2-deficient tumor organoids were more resistant to Silmitasertib-induced cell death, confirming that SALL2 sensitizes cancer cells to CK2 inhibition. We identified a novel CK2-dependent mechanism for SALL2 regulation and provided new insights into the interplay between these two proteins and their role in cell survival and proliferation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



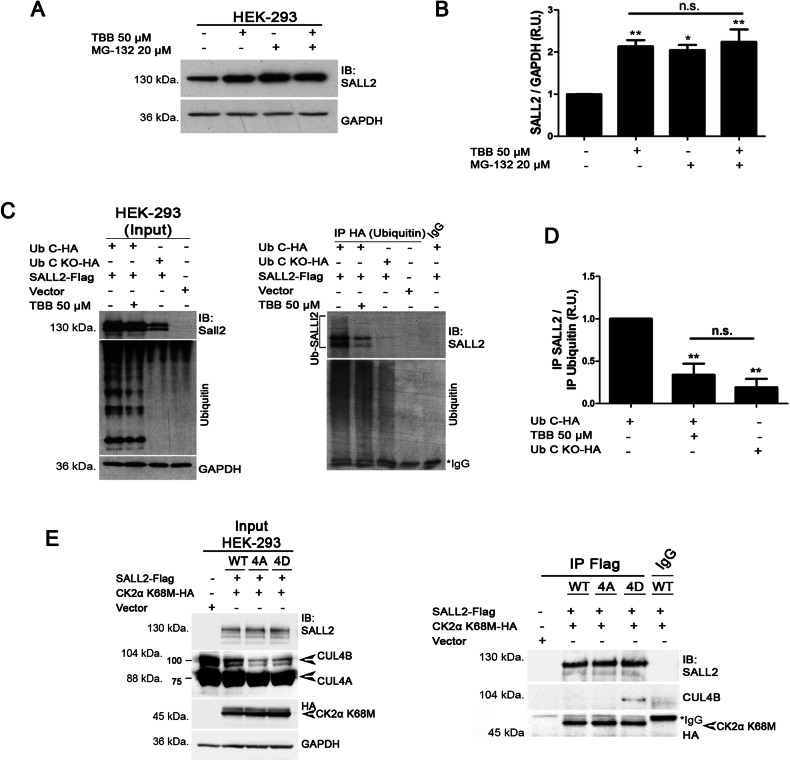


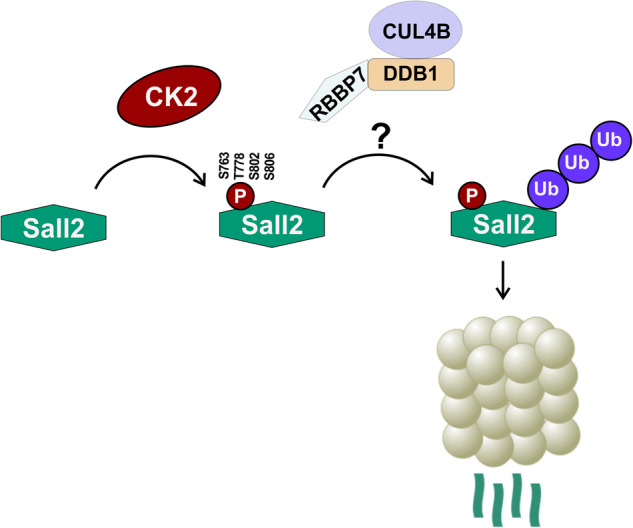
References
Publication types
MeSH terms
Substances
Grants and funding
- 1151031/Fondo Nacional de Desarrollo Científico y Tecnológico (National Fund for Scientific and Technological Development)
- 1191172/Fondo Nacional de Desarrollo Científico y Tecnológico (National Fund for Scientific and Technological Development)
- 1201215/Fondo Nacional de Desarrollo Científico y Tecnológico (National Fund for Scientific and Technological Development)
- 21120760/Consejo Nacional de Innovación, Ciencia y Tecnología (CONICYT)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
