

This is a preprint.
Nanopore sequencing of 1000 Genomes Project samples to build a comprehensive catalog of human genetic variation
- PMID: 38496498
- PMCID: PMC10942501
- DOI: 10.1101/2024.03.05.24303792
Nanopore sequencing of 1000 Genomes Project samples to build a comprehensive catalog of human genetic variation
Update in
-
High-coverage nanopore sequencing of samples from the 1000 Genomes Project to build a comprehensive catalog of human genetic variation.Genome Res. 2024 Nov 20;34(11):2061-2073. doi: 10.1101/gr.279273.124. Genome Res. 2024. PMID: 39358015 Free PMC article.
Abstract
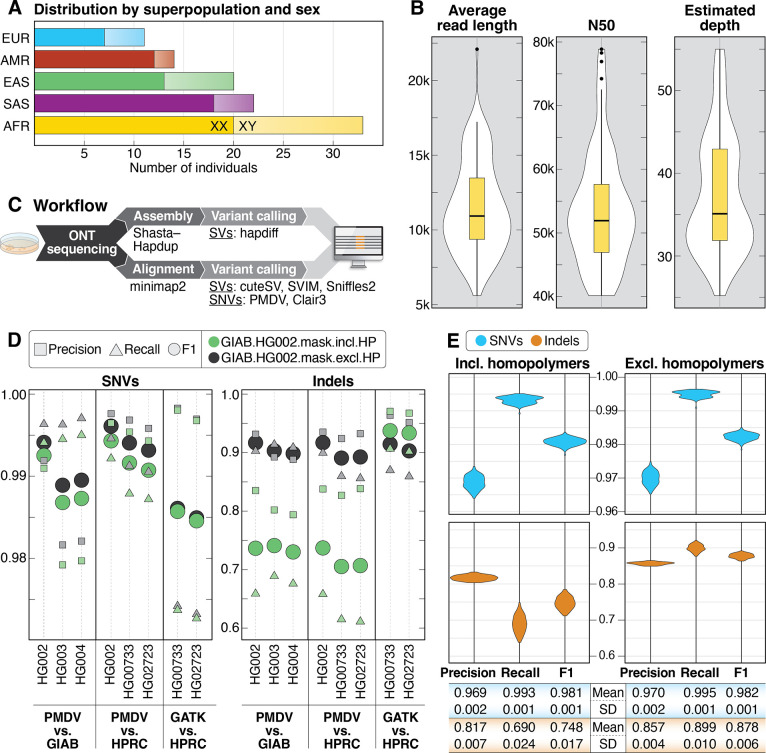
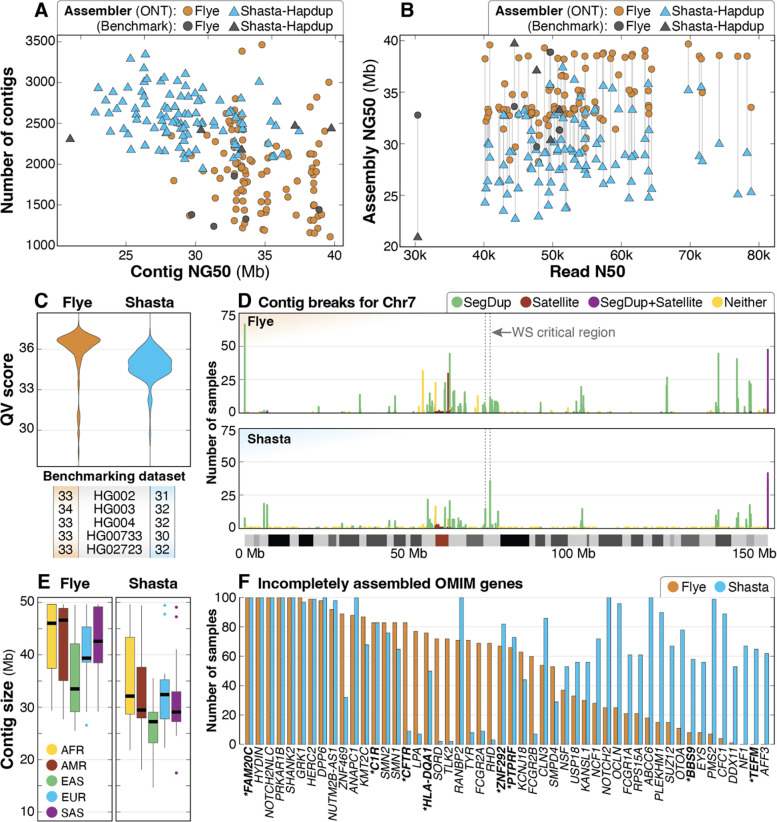
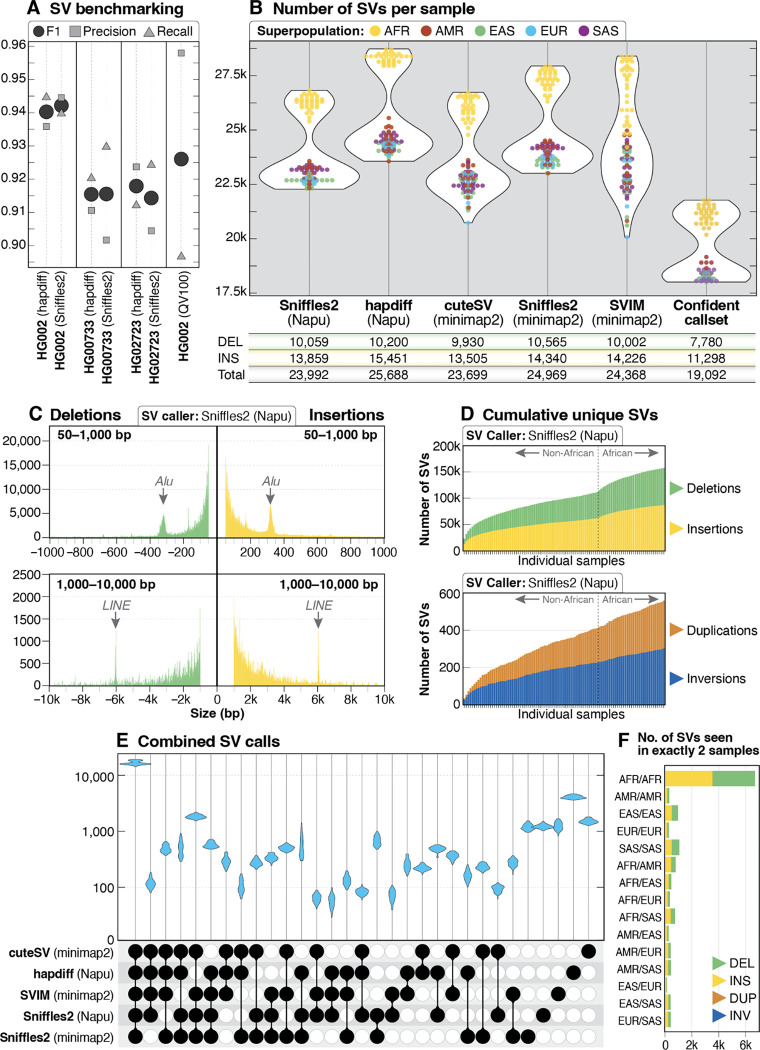
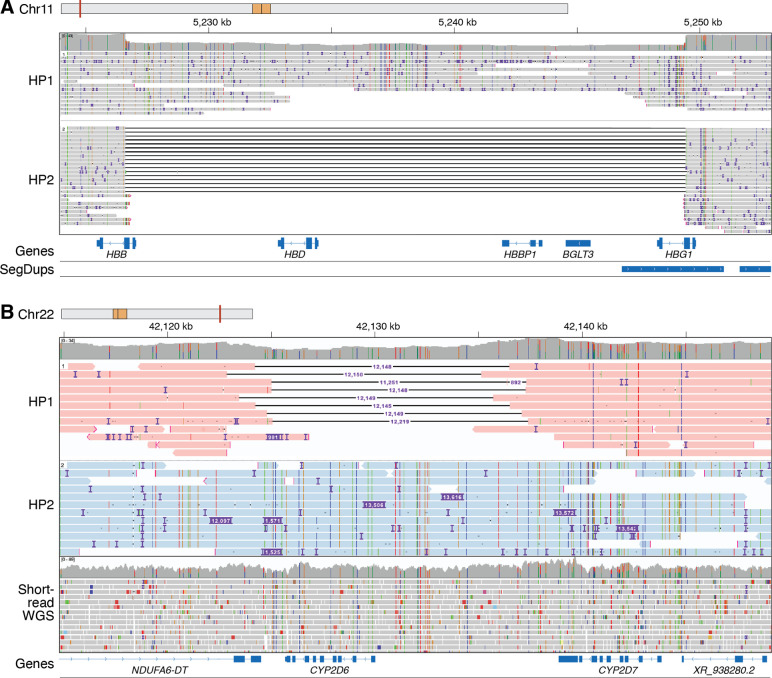
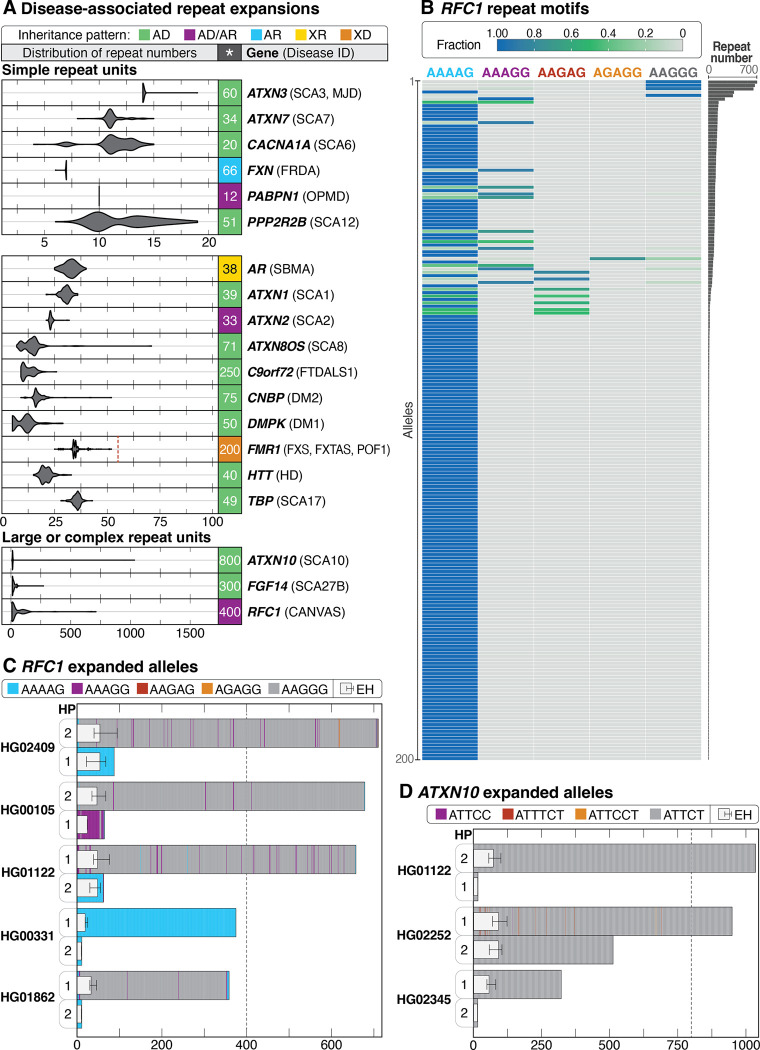
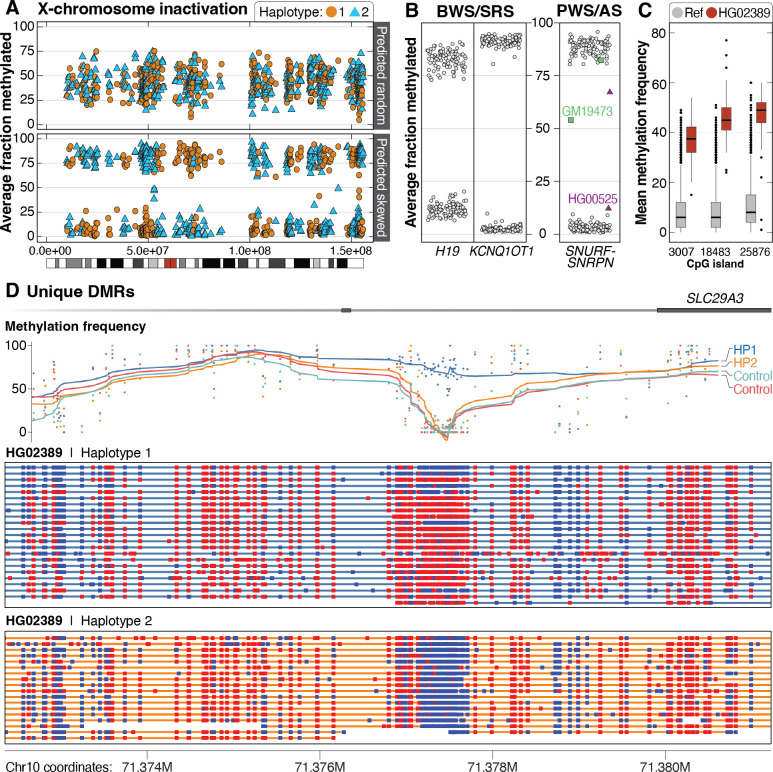
Less than half of individuals with a suspected Mendelian condition receive a precise molecular diagnosis after comprehensive clinical genetic testing. Improvements in data quality and costs have heightened interest in using long-read sequencing (LRS) to streamline clinical genomic testing, but the absence of control datasets for variant filtering and prioritization has made tertiary analysis of LRS data challenging. To address this, the 1000 Genomes Project ONT Sequencing Consortium aims to generate LRS data from at least 800 of the 1000 Genomes Project samples. Our goal is to use LRS to identify a broader spectrum of variation so we may improve our understanding of normal patterns of human variation. Here, we present data from analysis of the first 100 samples, representing all 5 superpopulations and 19 subpopulations. These samples, sequenced to an average depth of coverage of 37x and sequence read N50 of 54 kbp, have high concordance with previous studies for identifying single nucleotide and indel variants outside of homopolymer regions. Using multiple structural variant (SV) callers, we identify an average of 24,543 high-confidence SVs per genome, including shared and private SVs likely to disrupt gene function as well as pathogenic expansions within disease-associated repeats that were not detected using short reads. Evaluation of methylation signatures revealed expected patterns at known imprinted loci, samples with skewed X-inactivation patterns, and novel differentially methylated regions. All raw sequencing data, processed data, and summary statistics are publicly available, providing a valuable resource for the clinical genetics community to discover pathogenic SVs.
Keywords: 1000 Genomes Project; Nanopore sequencing; long-read sequencing; methylation; repeat expansions; structural variation.
Conflict of interest statement
COMPETING INTEREST STATEMENT WDC, ML, FS, and DEM have received research support and/or consumables from ONT. WDC, JG, FS, and DEM have received travel funding to speak on behalf of ONT. DEM is on a scientific advisory board at ONT. FS has received research support from Illumina, Genetech, and PacBio. SBM is an advisor to BioMarin, MyOme, and Tenaya Therapeutics. EEE is a scientific advisory board (SAB) member of Variant Bio, Inc. DEM holds stock options in MyOme.
Figures
References
-
- Alonso I, Jardim LB, Artigalas O, Saraiva-Pereira ML, Matsuura T, Ashizawa T, Sequeiros J, Silveira I. 2006. Reduced penetrance of intermediate size alleles in spinocerebellar ataxia type 10. Neurology 66: 1602–1604. - PubMed
Publication types
Grants and funding
- U24 HG010263/HG/NHGRI NIH HHS/United States
- R01 HG013017/HG/NHGRI NIH HHS/United States
- U01 AG058589/AG/NIA NIH HHS/United States
- R21 AI174130/AI/NIAID NIH HHS/United States
- U01 HG011744/HG/NHGRI NIH HHS/United States
- DP5 OD033357/OD/NIH HHS/United States
- K22 HG000044/HG/NHGRI NIH HHS/United States
- UG3 NS132105/NS/NINDS NIH HHS/United States
- U24 HG011746/HG/NHGRI NIH HHS/United States
- R03 CA272952/CA/NCI NIH HHS/United States
- T32 HG000035/HG/NHGRI NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- U01 CA253481/CA/NCI NIH HHS/United States
- U01 HG011745/HG/NHGRI NIH HHS/United States
- R50 CA243890/CA/NCI NIH HHS/United States
- U01 HG011762/HG/NHGRI NIH HHS/United States
- T32 HG000044/HG/NHGRI NIH HHS/United States
- U01 HG011755/HG/NHGRI NIH HHS/United States
- U01 HG011758/HG/NHGRI NIH HHS/United States
- U01 DA057530/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources