

This is a preprint.
Novel syndromic neurodevelopmental disorder caused by de novo deletion of CHASERR, a long noncoding RNA
- PMID: 38496558
- PMCID: PMC10942497
- DOI: 10.1101/2024.01.31.24301497
Novel syndromic neurodevelopmental disorder caused by de novo deletion of CHASERR, a long noncoding RNA
Abstract
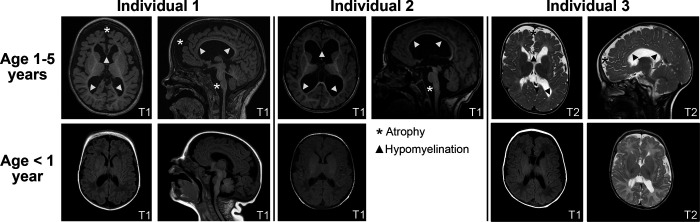
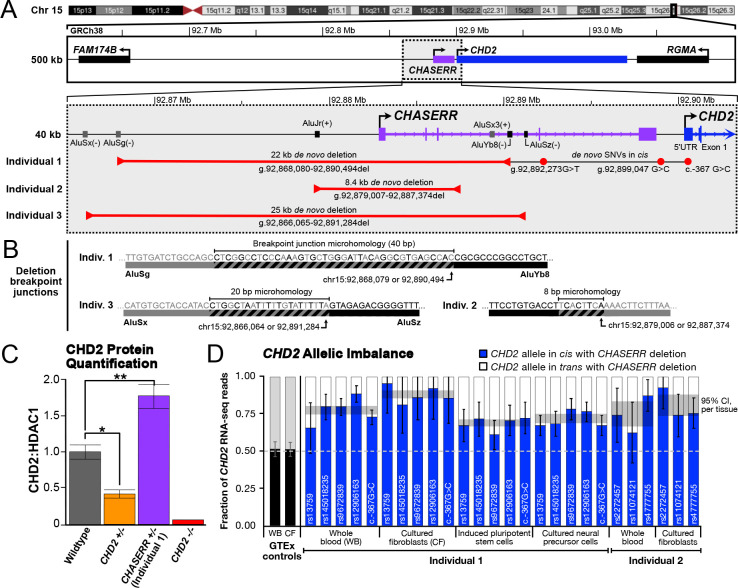
Genes encoding long non-coding RNAs (lncRNAs) comprise a large fraction of the human genome, yet haploinsufficiency of a lncRNA has not been shown to cause a Mendelian disease. CHASERR is a highly conserved human lncRNA adjacent to CHD2-a coding gene in which de novo loss-of-function variants cause developmental and epileptic encephalopathy. Here we report three unrelated individuals each harboring an ultra-rare heterozygous de novo deletion in the CHASERR locus. We report similarities in severe developmental delay, facial dysmorphisms, and cerebral dysmyelination in these individuals, distinguishing them from the phenotypic spectrum of CHD2 haploinsufficiency. We demonstrate reduced CHASERR mRNA expression and corresponding increased CHD2 mRNA and protein in whole blood and patient-derived cell lines-specifically increased expression of the CHD2 allele in cis with the CHASERR deletion, as predicted from a prior mouse model of Chaserr haploinsufficiency. We show for the first time that de novo structural variants facilitated by Alu-mediated non-allelic homologous recombination led to deletion of a non-coding element (the lncRNA CHASERR) to cause a rare syndromic neurodevelopmental disorder. We also demonstrate that CHD2 has bidirectional dosage sensitivity in human disease. This work highlights the need to carefully evaluate other lncRNAs, particularly those upstream of genes associated with Mendelian disorders.
Figures
Similar articles
-
Neurodevelopmental Disorder Caused by Deletion of CHASERR, a lncRNA Gene.N Engl J Med. 2024 Oct 24;391(16):1511-1518. doi: 10.1056/NEJMoa2400718. N Engl J Med. 2024. PMID: 39442041 Free PMC article.
-
A roadmap to cure CHD2-related disorders.Ther Adv Rare Dis. 2024 Oct 8;5:26330040241283749. doi: 10.1177/26330040241283749. eCollection 2024 Jan-Dec. Ther Adv Rare Dis. 2024. PMID: 39391213 Free PMC article. Review.
-
Regulation of CHD2 expression by the Chaserr long noncoding RNA gene is essential for viability.Nat Commun. 2019 Nov 8;10(1):5092. doi: 10.1038/s41467-019-13075-8. Nat Commun. 2019. PMID: 31704914 Free PMC article.
-
m6A-Mediated Upregulation of lncRNA CHASERR Promotes the Progression of Glioma by Modulating the miR-6893-3p/TRIM14 Axis.Mol Neurobiol. 2024 Aug;61(8):5418-5440. doi: 10.1007/s12035-023-03911-w. Epub 2024 Jan 9. Mol Neurobiol. 2024. PMID: 38193984
-
Transcription regulation by long non-coding RNAs: mechanisms and disease relevance.Nat Rev Mol Cell Biol. 2024 May;25(5):396-415. doi: 10.1038/s41580-023-00694-9. Epub 2024 Jan 19. Nat Rev Mol Cell Biol. 2024. PMID: 38242953 Free PMC article. Review.
Cited by
-
A positive relationship between weight-adjusted waist index and non-alcoholic fatty liver disease: a study on US adolescents.Front Med (Lausanne). 2025 Jan 7;11:1424667. doi: 10.3389/fmed.2024.1424667. eCollection 2024. Front Med (Lausanne). 2025. PMID: 39845834 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources