

Aberrant DNA methylation distorts developmental trajectories in atypical teratoid/rhabdoid tumors
- PMID: 38499326
- PMCID: PMC10948937
- DOI: 10.26508/lsa.202302088
Aberrant DNA methylation distorts developmental trajectories in atypical teratoid/rhabdoid tumors
Abstract
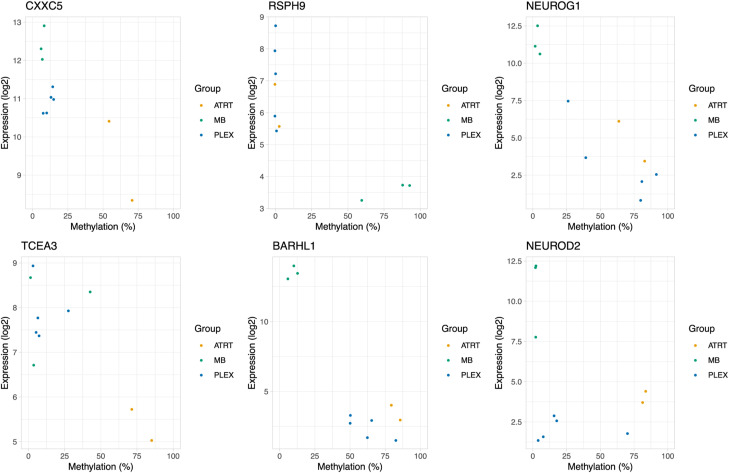
Atypical teratoid/rhabdoid tumors (AT/RTs) are pediatric brain tumors known for their aggressiveness and aberrant but still unresolved epigenetic regulation. To better understand their malignancy, we investigated how AT/RT-specific DNA hypermethylation was associated with gene expression and altered transcription factor binding and how it is linked to upstream regulation. Medulloblastomas, choroid plexus tumors, pluripotent stem cells, and fetal brain were used as references. A part of the genomic regions, which were hypermethylated in AT/RTs similarly as in pluripotent stem cells and demethylated in the fetal brain, were targeted by neural transcriptional regulators. AT/RT-unique DNA hypermethylation was associated with polycomb repressive complex 2 and linked to suppressed genes with a role in neural development and tumorigenesis. Activity of the several NEUROG/NEUROD pioneer factors, which are unable to bind to methylated DNA, was compromised via the suppressed expression or DNA hypermethylation of their target sites, which was also experimentally validated for NEUROD1 in medulloblastomas and AT/RT samples. These results highlight and characterize the role of DNA hypermethylation in AT/RT malignancy and halted neural cell differentiation.
© 2024 Pekkarinen et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


















Similar articles
-
Development and epigenetic regulation of Atypical teratoid/rhabdoid tumors in the context of cell-of-origin and halted cell differentiation.Neurooncol Adv. 2024 Sep 23;6(1):vdae162. doi: 10.1093/noajnl/vdae162. eCollection 2024 Jan-Dec. Neurooncol Adv. 2024. PMID: 39465218 Free PMC article. Review.
-
Alpha-internexin expression in medulloblastomas and atypical teratoid-rhabdoid tumors.Clin Neuropathol. 2003 Sep-Oct;22(5):215-21. Clin Neuropathol. 2003. PMID: 14531545
-
Integrated genomics has identified a new AT/RT-like yet INI1-positive brain tumor subtype among primary pediatric embryonal tumors.BMC Med Genomics. 2015 Jun 25;8:32. doi: 10.1186/s12920-015-0103-3. BMC Med Genomics. 2015. PMID: 26109171 Free PMC article.
-
Epigenetic repression of RASSF1A but not CASP8 in supratentorial PNET (sPNET) and atypical teratoid/rhabdoid tumors (AT/RT) of childhood.Oncogene. 2006 Feb 16;25(7):1111-7. doi: 10.1038/sj.onc.1209137. Oncogene. 2006. PMID: 16186793
-
Advances in the molecular classification of pediatric brain tumors: a guide to the galaxy.J Pathol. 2020 Jul;251(3):249-261. doi: 10.1002/path.5457. Epub 2020 Jun 10. J Pathol. 2020. PMID: 32391583 Review.
Cited by
-
Metastasis of colon cancer requires Dickkopf-2 to generate cancer cells with Paneth cell properties.Elife. 2024 Nov 13;13:RP97279. doi: 10.7554/eLife.97279. Elife. 2024. PMID: 39535280 Free PMC article.
-
Development and epigenetic regulation of Atypical teratoid/rhabdoid tumors in the context of cell-of-origin and halted cell differentiation.Neurooncol Adv. 2024 Sep 23;6(1):vdae162. doi: 10.1093/noajnl/vdae162. eCollection 2024 Jan-Dec. Neurooncol Adv. 2024. PMID: 39465218 Free PMC article. Review.
-
Epigenetic modifications and their roles in pediatric brain tumor formation: emerging insights from chromatin dysregulation.Front Oncol. 2025 Jun 17;15:1569548. doi: 10.3389/fonc.2025.1569548. eCollection 2025. Front Oncol. 2025. PMID: 40599851 Free PMC article. Review.
-
SMARCB1-related schwannomatosis and other SMARCB1-associated phenotypes: clinical spectrum and molecular pathogenesis.Fam Cancer. 2025 Aug 12;24(3):64. doi: 10.1007/s10689-025-00486-4. Fam Cancer. 2025. PMID: 40794298 Free PMC article. Review.
References
-
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodological) 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases