

Case report: Rapidly progressive neurocognitive disorder with a fatal outcome in a patient with PU.1 mutated agammaglobulinemia
- PMID: 38500873
- PMCID: PMC10945545
- DOI: 10.3389/fimmu.2024.1324679
Case report: Rapidly progressive neurocognitive disorder with a fatal outcome in a patient with PU.1 mutated agammaglobulinemia
Abstract
Introduction: PU.1-mutated agammaglobulinemia (PU.MA) represents a recently described autosomal-dominant form of agammaglobulinemia caused by mutation of the SPI1 gene. This gene codes for PU.1 pioneer transcription factor important for the maturation of monocytes, B lymphocytes, and conventional dendritic cells. Only six cases with PU.MA, presenting with chronic sinopulmonary and systemic enteroviral infections, have been previously described. Accumulating literature evidence suggests a possible relationship between SPI1 mutation, microglial phagocytic dysfunction, and the development of Alzheimer's disease (AD).
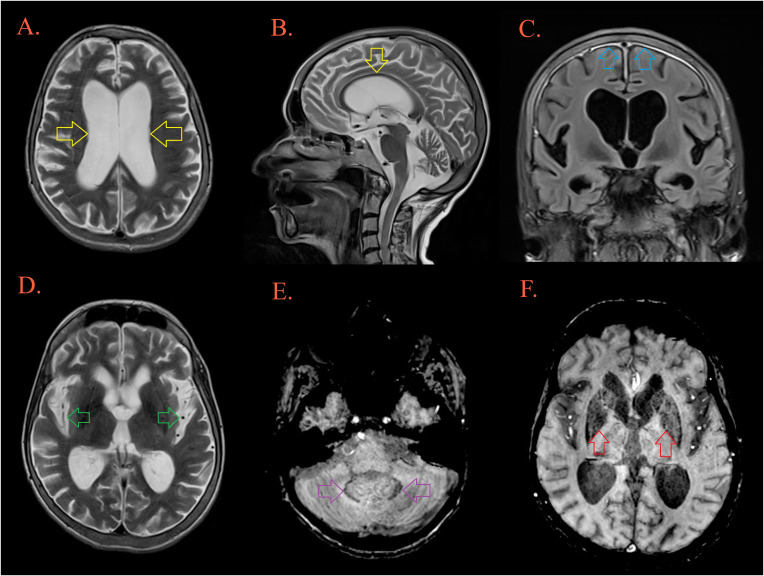
Case description: We present a Caucasian female patient born from a non-consanguineous marriage, who was diagnosed with agammaglobulinemia at the age of 15 years when the immunoglobulin replacement therapy was started. During the following seventeen years, she was treated for recurrent respiratory and intestinal infections. At the age of 33 years, the diagnosis of celiac-like disease was established. Five years later progressive cognitive deterioration, unstable gait, speech disturbances, and behavioral changes developed. Comprehensive microbiological investigations were negative, excluding possible infective etiology. Brain MRI, 18FDG-PET-CT, and neuropsychological testing were suggestive for a diagnosis of a frontal variant of AD. Clinical exome sequencing revealed the presence of a novel frameshift heterozygous variant c.441dup in exon 4 of the SPI1 gene. Despite intensive therapy, the patient passed away a few months after the onset of the first neurological symptoms.
Conclusion: We describe the first case of PU.MA patient presenting with a rapidly progressive neurocognitive deterioration. The possible role of microglial dysfunction in patients with SPI1 mutation could explain their susceptibility to neurodegenerative diseases thus highlighting the importance of genetic testing in patients with inborn errors of immunity. Since PU.MA represents a newly described form of agammaglobulinemia, our case expands the spectrum of manifestations associated with SPI1 mutation.
Keywords: Alzheimer’s disease; PU.1; SPI1; agammaglobulinemia; exome sequencing; neurocognitive disorders.
Copyright © 2024 Miskovic, Ljubicic, Bonaci-Nikolic, Petkovic, Markovic, Rankovic, Djordjevic, Stankovic, Klaassen, Pavlovic and Stojanovic.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures




References
-
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. . Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2022) 42:1473–507. doi: 10.1007/s10875-022-01289-3 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical