

Exemestane plus everolimus and palbociclib in metastatic breast cancer: clinical response and genomic/transcriptomic determinants of resistance in a phase I/II trial
- PMID: 38503755
- PMCID: PMC10951222
- DOI: 10.1038/s41467-024-45835-6
Exemestane plus everolimus and palbociclib in metastatic breast cancer: clinical response and genomic/transcriptomic determinants of resistance in a phase I/II trial
Abstract
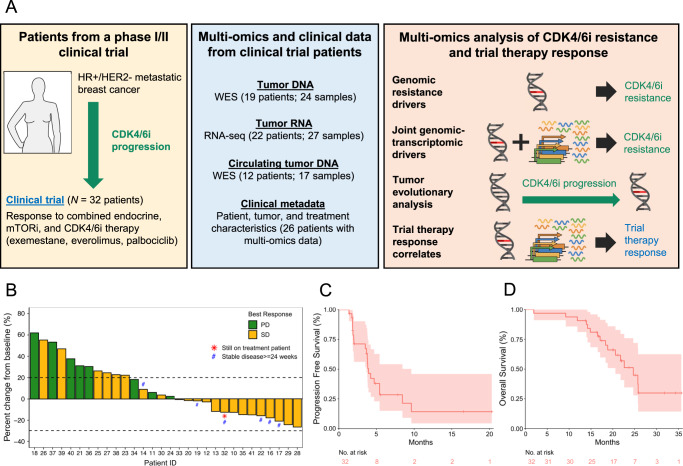
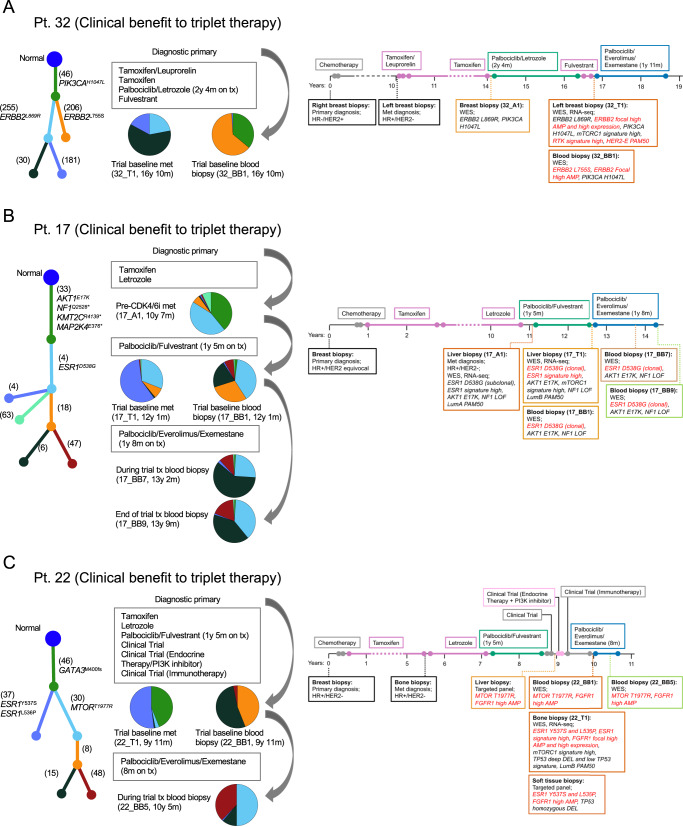
The landscape of cyclin-dependent kinase 4/6 inhibitor (CDK4/6i) resistance is still being elucidated and the optimal subsequent therapy to overcome resistance remains uncertain. Here we present the final results of a phase Ib/IIa, open-label trial (NCT02871791) of exemestane plus everolimus and palbociclib for CDK4/6i-resistant metastatic breast cancer. The primary objective of phase Ib was to evaluate safety and tolerability and determine the maximum tolerated dose/recommended phase II dose (100 mg palbociclib, 5 mg everolimus, 25 mg exemestane). The primary objective of phase IIa was to determine the clinical benefit rate (18.8%, n = 6/32), which did not meet the predefined endpoint (65%). Secondary objectives included pharmacokinetic profiling (phase Ib), objective response rate, disease control rate, duration of response, and progression free survival (phase IIa), and correlative multi-omics analysis to investigate biomarkers of resistance to CDK4/6i. All participants were female. Multi-omics data from the phase IIa patients (n = 24 tumor/17 blood biopsy exomes; n = 27 tumor transcriptomes) showed potential mechanisms of resistance (convergent evolution of HER2 activation, BRAFV600E), identified joint genomic/transcriptomic resistance features (ESR1 mutations, high estrogen receptor pathway activity, and a Luminal A/B subtype; ERBB2/BRAF mutations, high RTK/MAPK pathway activity, and a HER2-E subtype), and provided hypothesis-generating results suggesting that mTOR pathway activation correlates with response to the trial's therapy. Our results illustrate how genome and transcriptome sequencing may help better identify patients likely to respond to CDK4/6i therapies.
© 2024. The Author(s).
Conflict of interest statement
J.G. owns stocks in the biotechnology exchange-traded funds CNCR, IDNA, IBB, and XBI, and owned stocks in Adaptive Biotechnologies, 2seventy bio, and bluebird bio. R.B.-S. has received consulting fees from AstraZeneca, Eli Lilly, Libbs, Merck, Roche, and Zodiac; non-CME fees from Bard Access, BMS, Eli Lilly, Libbs, Merck, Novartis, Pfizer, and Roche; has carried out contracted research for Roche; and has received travel, accommodation and expenses from Eli Lilly, Roche, Daichi Sankyo, and Merck. E.J. is a current employee of Repare Therapeutics. J.E.B.-B. is a current employee of Cellarity. A.R.F. has received honoraria from Bayer, Daiichi Sankyo, Novartis, and Roche; and has received travel grants from Roche. N.U.L. has received consulting fees from Puma, Seattle Genetics, Daichii-Sankyo, AstraZeneca, Denali Therapeutics, Prelude Therapeutics, Olema Pharmaceuticals, Aleta BioPharma, Affinia Therapeutics, Voyager Therapeutics, Janssen, and Blueprint Medicines; has received institutional research support from Genentech, Pfizer, Merck, Seattle Genetics, Zion Pharmaceuticals, Olema Pharmaceuticals, and AstraZeneca; and has stocks and other ownership interests in Artera Inc. (<$50k and <5% as it relates to consulting activities—options are not currently valued or in-hand); and royalties from UptoDate (book). N.W. is a current employee of Genentech; has been a consultant/advisor for Eli Lilly; has been in the scientific advisory board for Relay Therapeutics and Flare Therapeutics; has received grant support from AstraZeneca and Puma Biotechnologies. S.M.T. reports consulting or advisory roles for Novartis, Pfizer, Merck, Eli Lilly, AstraZeneca, Genentech/Roche, Eisai, Sanofi, Bristol Myers Squibb, Seattle Genetics, CytomX Therapeutics, Daiichi Sankyo, Gilead, Ellipses Pharma, 4D Pharma, OncoSec Medical Inc., BeyondSpring Pharmaceuticals, OncXerna, Zymeworks, Zentalis, Blueprint Medicines, Reveal Genomics, ARC Therapeutics, Infinity Therapeutics, Myovant, Zetagen, Umoja Biopharma, Artios Pharma, Menarini/Stemline, Aadi Biopharma, Bayer, and Incyte Corp; and research funding from Genentech/Roche, Merck, Exelixis, Pfizer, Lilly, Novartis, Bristol Myers Squibb, Eisai, AstraZeneca, Gilead, NanoString Technologies, Gilead, Seattle Genetics, and OncoPep. The remaining authors declare no competing interests.
Figures





References
-
- O’Leary B, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov. 2018;8:1390–1403. doi: 10.1158/2159-8290.CD-18-0264. - DOI - PMC - PubMed
-
- Wander SA, et al. The genomic landscape of intrinsic and acquired resistance to cyclin-dependent kinase 4/6 inhibitors in patients with hormone receptor–positive metastatic breast cancer. Cancer Discov. 2020;10:1174–1193. doi: 10.1158/2159-8290.CD-19-1390. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
