

Design of amyloidogenic peptide traps
- PMID: 38503834
- PMCID: PMC11288891
- DOI: 10.1038/s41589-024-01578-5
Design of amyloidogenic peptide traps
Abstract
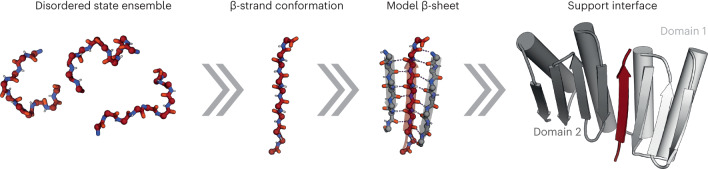
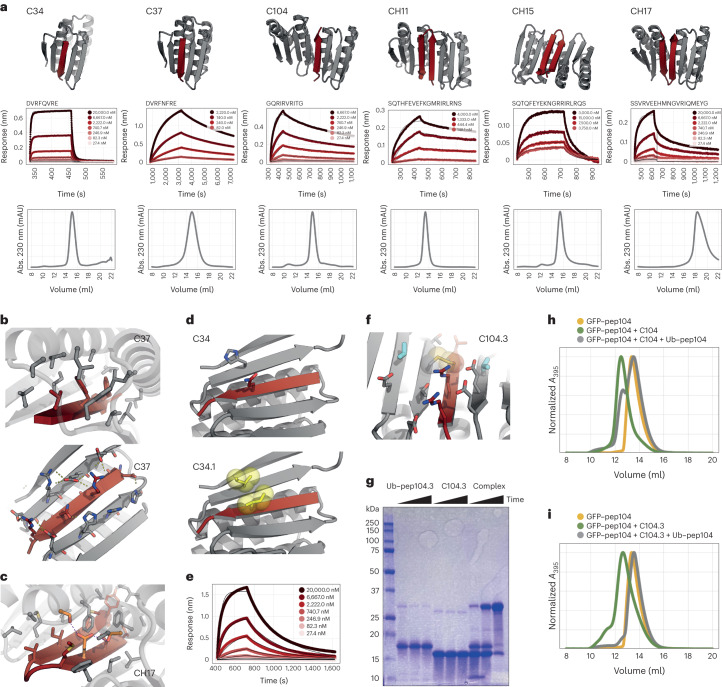
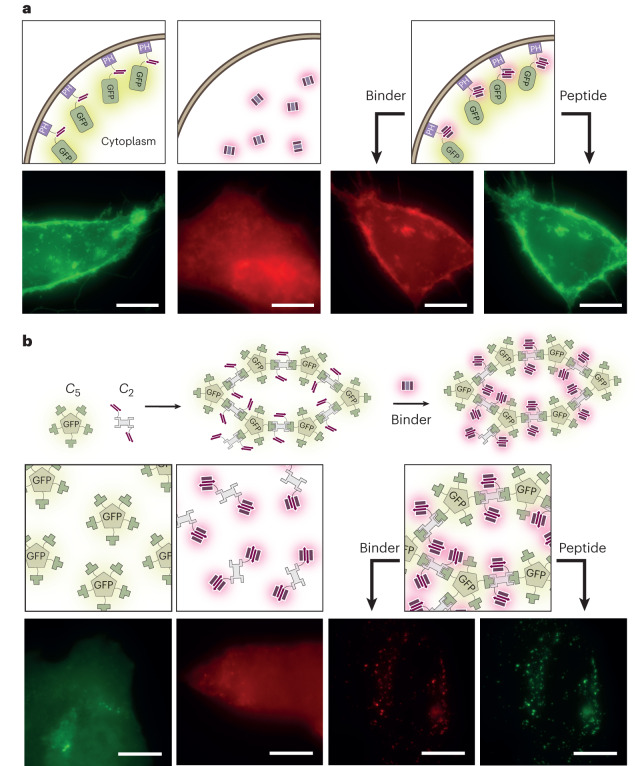
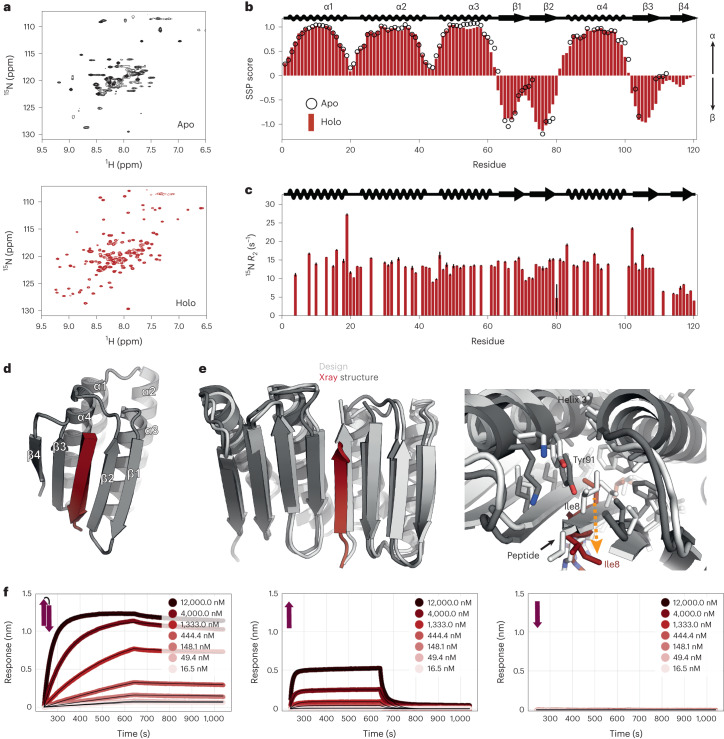
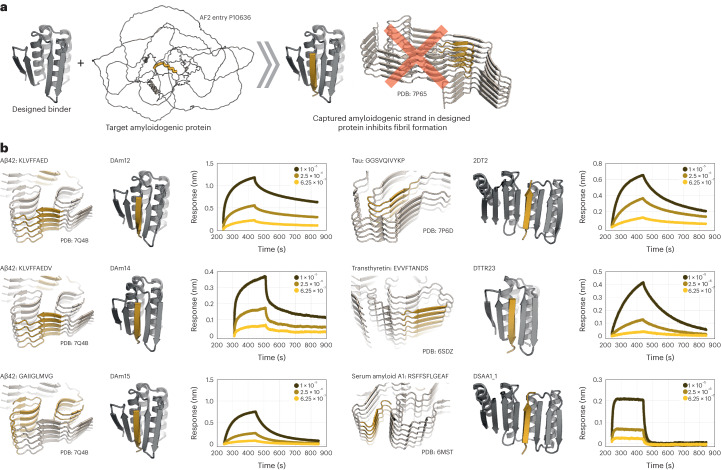
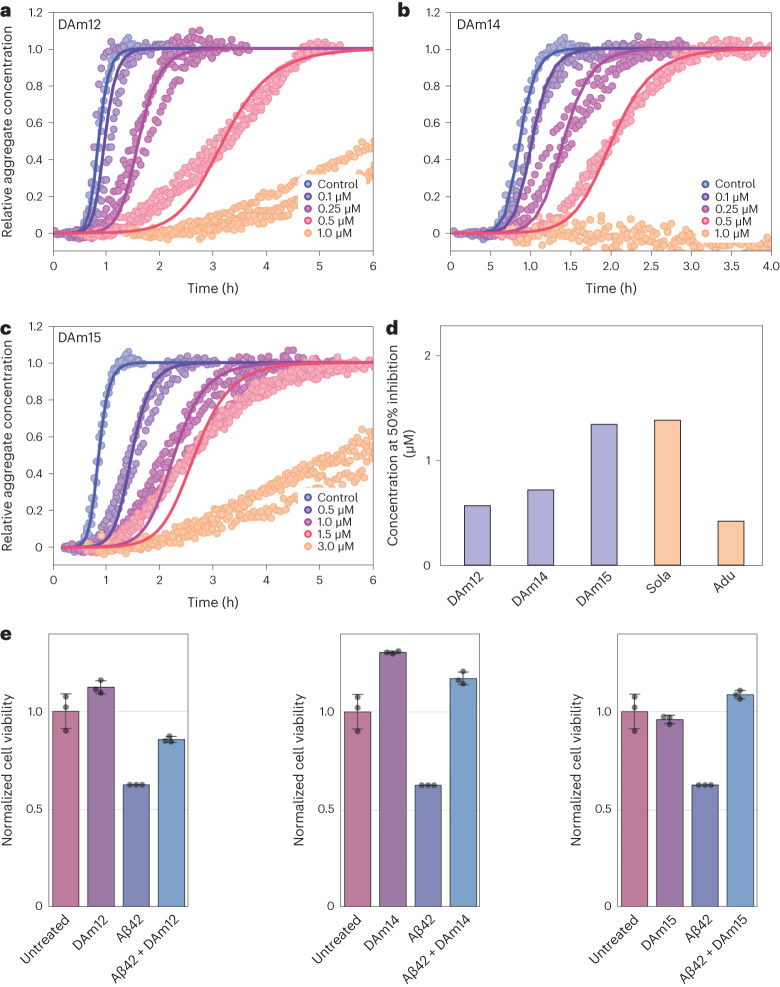
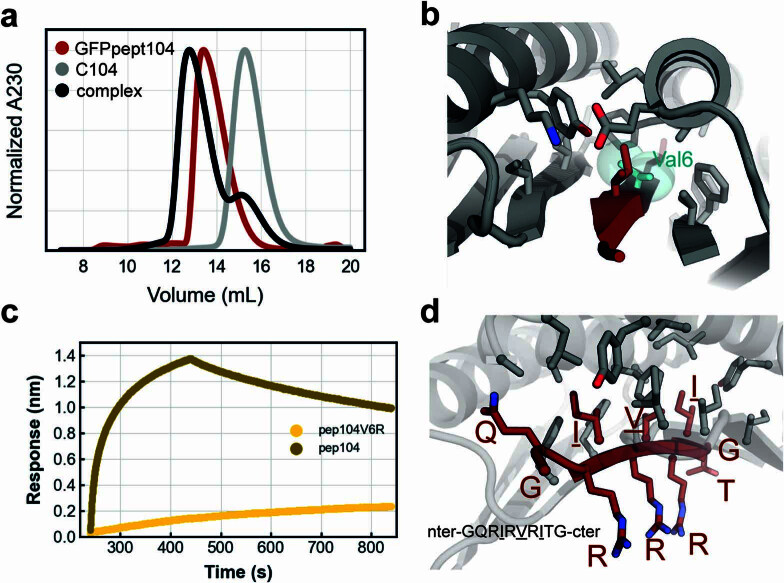
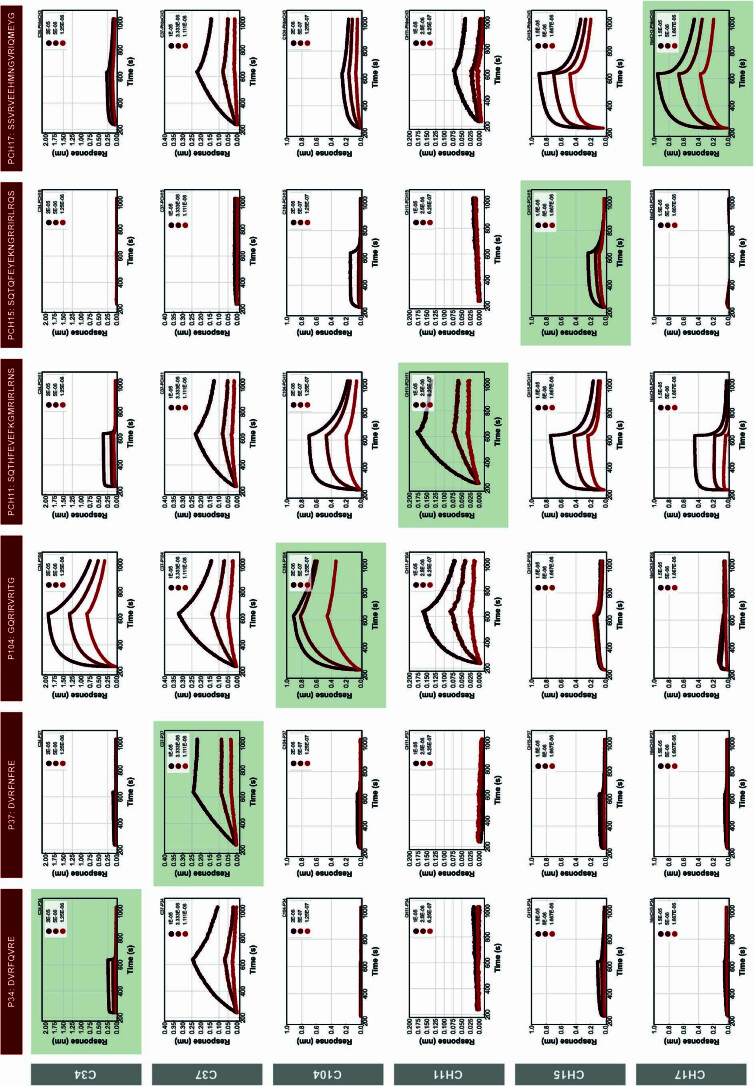
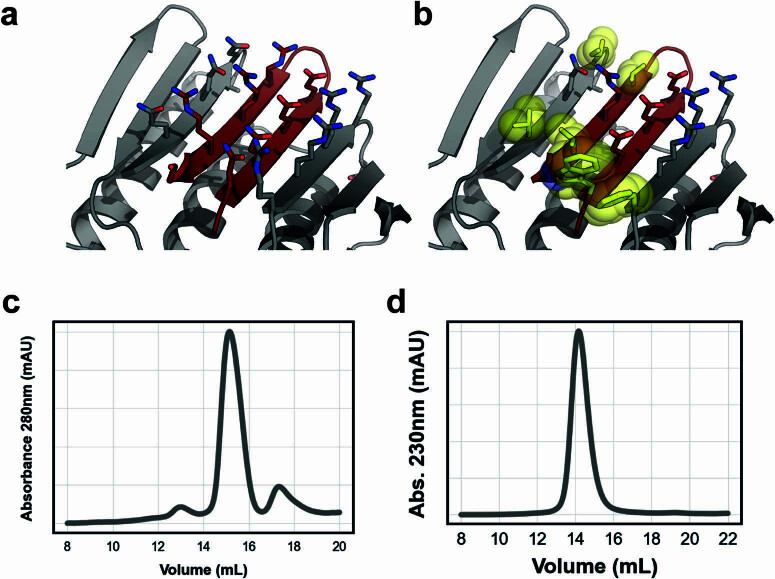
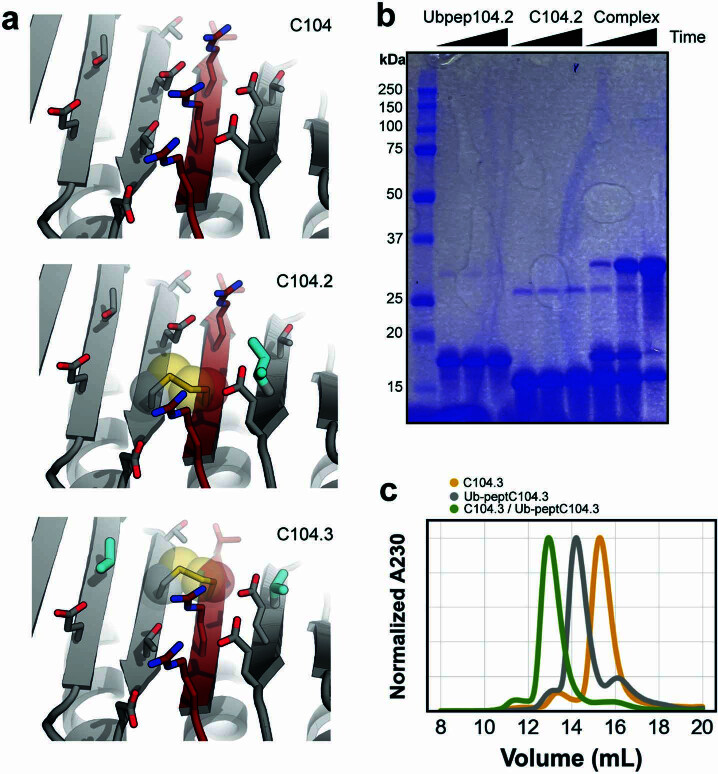
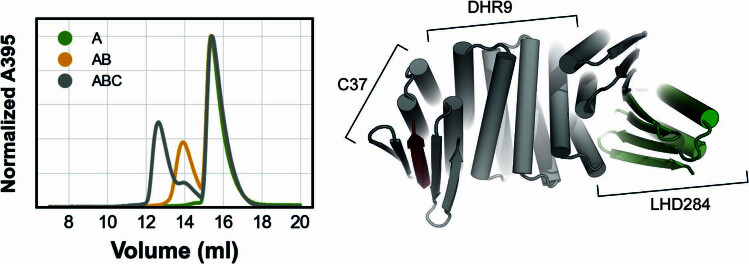
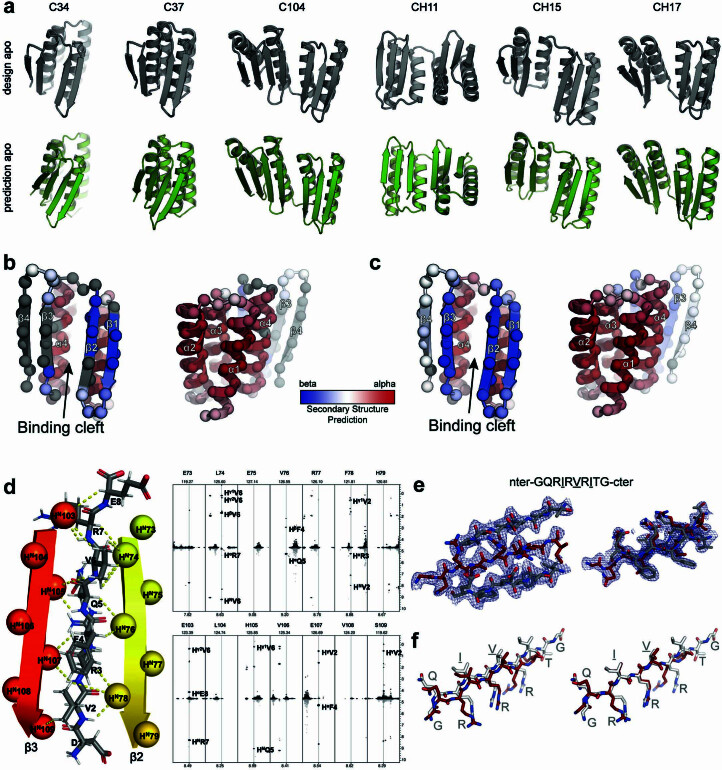
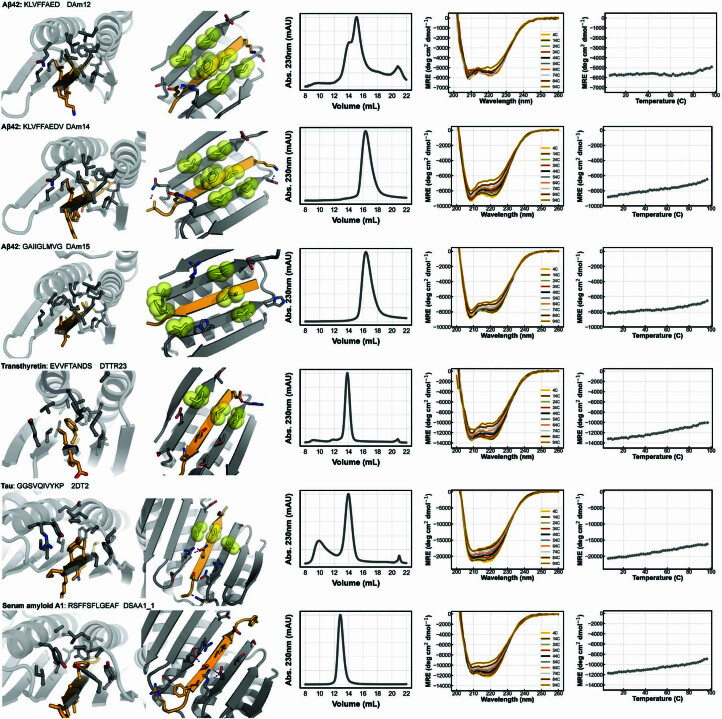
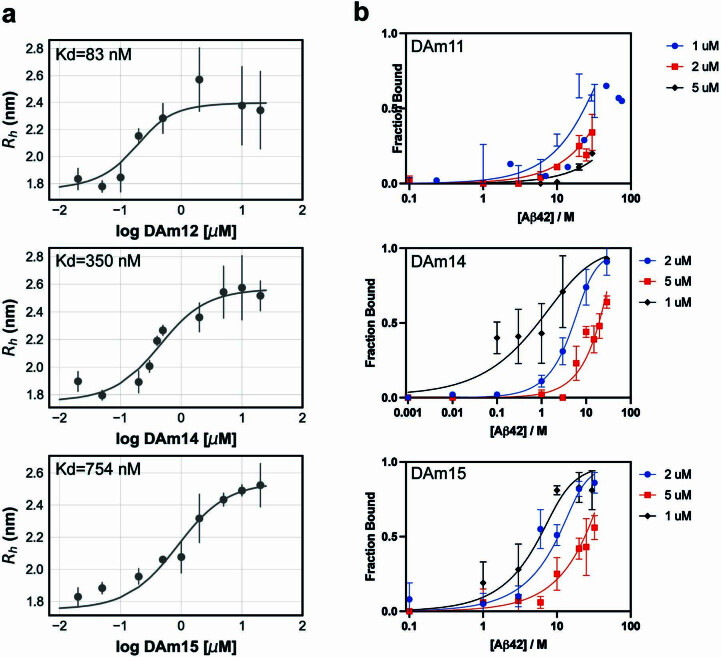
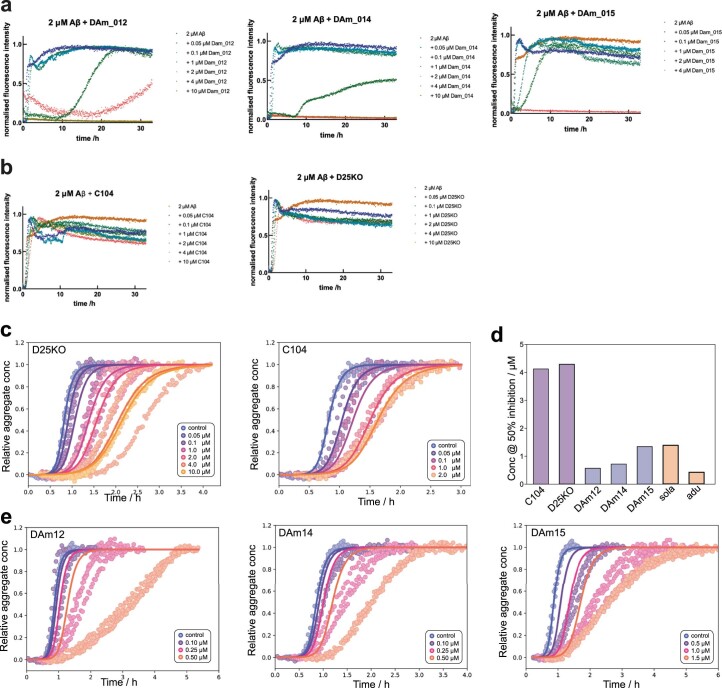
Segments of proteins with high β-strand propensity can self-associate to form amyloid fibrils implicated in many diseases. We describe a general approach to bind such segments in β-strand and β-hairpin conformations using de novo designed scaffolds that contain deep peptide-binding clefts. The designs bind their cognate peptides in vitro with nanomolar affinities. The crystal structure of a designed protein-peptide complex is close to the design model, and NMR characterization reveals how the peptide-binding cleft is protected in the apo state. We use the approach to design binders to the amyloid-forming proteins transthyretin, tau, serum amyloid A1 and amyloid β1-42 (Aβ42). The Aβ binders block the assembly of Aβ fibrils as effectively as the most potent of the clinically tested antibodies to date and protect cells from toxic Aβ42 species.
© 2024. The Author(s).
Conflict of interest statement
D.D.S., H.L.H. and D.B. are inventors on a PCT patent (PCT/US2024/010806) filed by the University of Washington that covers the use of the designed proteins and their variants described in this work. The remaining authors declare no competing interests.
Figures
References
-
- Tsai, C. J., Xu, D. & Nussinov, R. Protein folding via binding and vice versa. Fold. Des.3, R71–R80 (1998). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
