

Multicenter integrated analysis of noncoding CRISPRi screens
- PMID: 38504114
- PMCID: PMC11009116
- DOI: 10.1038/s41592-024-02216-7
Multicenter integrated analysis of noncoding CRISPRi screens
Abstract
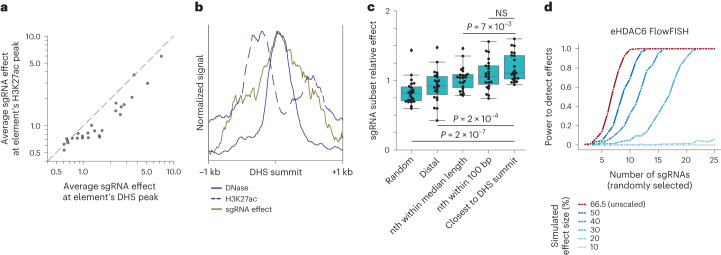
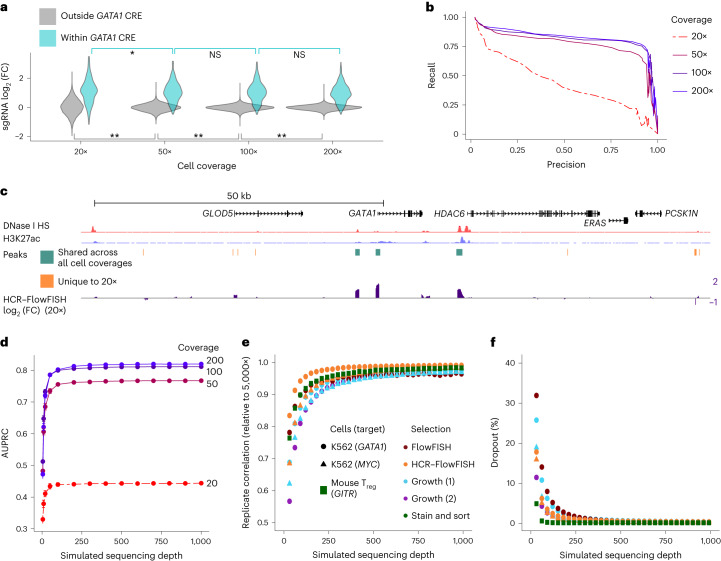
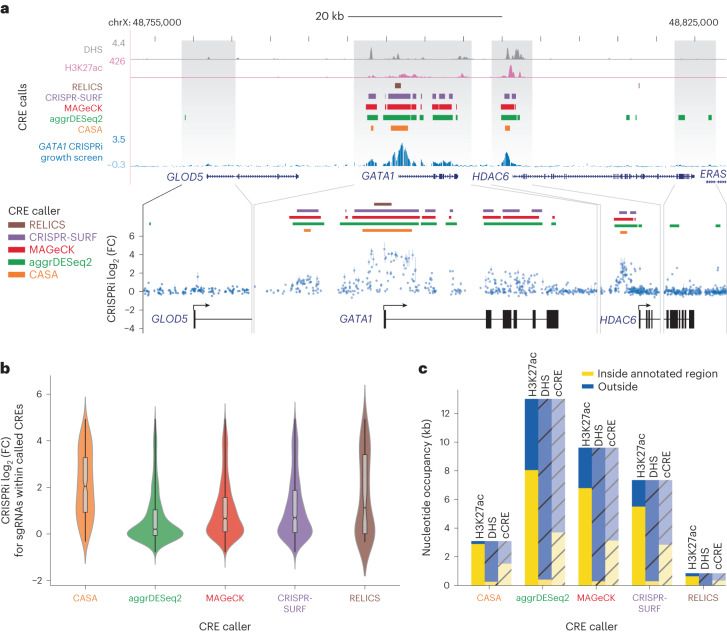
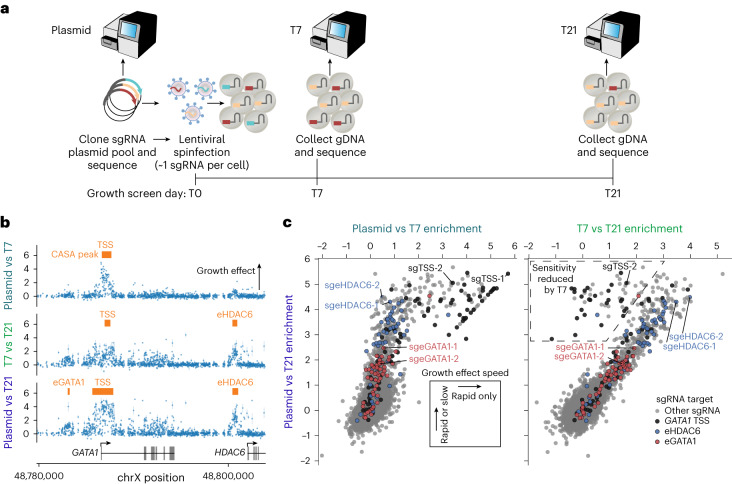
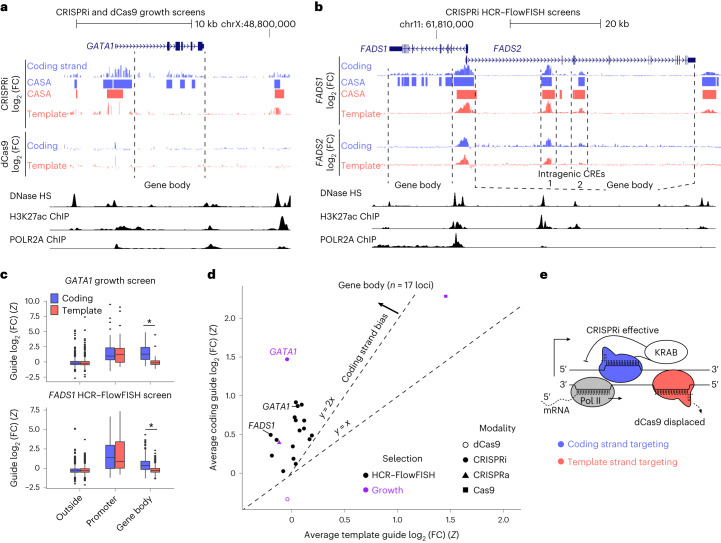
The ENCODE Consortium's efforts to annotate noncoding cis-regulatory elements (CREs) have advanced our understanding of gene regulatory landscapes. Pooled, noncoding CRISPR screens offer a systematic approach to investigate cis-regulatory mechanisms. The ENCODE4 Functional Characterization Centers conducted 108 screens in human cell lines, comprising >540,000 perturbations across 24.85 megabases of the genome. Using 332 functionally confirmed CRE-gene links in K562 cells, we established guidelines for screening endogenous noncoding elements with CRISPR interference (CRISPRi), including accurate detection of CREs that exhibit variable, often low, transcriptional effects. Benchmarking five screen analysis tools, we find that CASA produces the most conservative CRE calls and is robust to artifacts of low-specificity single guide RNAs. We uncover a subtle DNA strand bias for CRISPRi in transcribed regions with implications for screen design and analysis. Together, we provide an accessible data resource, predesigned single guide RNAs for targeting 3,275,697 ENCODE SCREEN candidate CREs with CRISPRi and screening guidelines to accelerate functional characterization of the noncoding genome.
© 2024. The Author(s).
Conflict of interest statement
A.K. is a scientific cofounder of Ravel Biotechnology, is on the scientific advisory board of PatchBio, SerImmune, AINovo, TensorBio and OpenTargets, is a consultant with Illumina and owns shares in DeepGenomics, Immuni and Freenome. C.A.G. is a cofounder of Tune Therapeutics and Locus Biosciences and is an advisor to Tune Therapeutics and Sarepta Therapeutics. C.A.G. is an inventor on patents and patent applications related to CRISPR epigenome editing. J.T. and M.C.B. acknowledge an outside interest in Stylus Medicine. L.L. is currently employed by Sana Biotechnology. D.Y. is currently employed by Amber Bio. P.C.S. is a cofounder of and consultant to Sherlock Biosciences and board member of Danaher Corporation. P.C.S. is a shareholder in both companies. W.J.G. is a cofounder of Epinomics and an advisor to 10x Genomics, Guardant Health and Centrillion. J.M.E. is an inventor on patents and patent applications related to CRISPR screening technologies, received materials from 10x Genomics unrelated to this study, and received speaking honoraria from GSK plc. The remaining authors declare no competing interests.
Figures










References
MeSH terms
Substances
Grants and funding
- UM1 HG012053/HG/NHGRI NIH HHS/United States
- R35 HG011329/HG/NHGRI NIH HHS/United States
- U01 HG012103/HG/NHGRI NIH HHS/United States
- UM1 HG009402/HG/NHGRI NIH HHS/United States
- R01 HG010741/HG/NHGRI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- R00 HG010669/HG/NHGRI NIH HHS/United States
- R01 NS128028/NS/NINDS NIH HHS/United States
- RM1 HG011123/HG/NHGRI NIH HHS/United States
- UM1 HG009428/HG/NHGRI NIH HHS/United States
- U24 HG009397/HG/NHGRI NIH HHS/United States
- R01 MH125236/MH/NIMH NIH HHS/United States
- T32 GM144291/GM/NIGMS NIH HHS/United States
- U01 HG009380/HG/NHGRI NIH HHS/United States
- U01 HG009395/HG/NHGRI NIH HHS/United States
- U01 HG009431/HG/NHGRI NIH HHS/United States
- K00 DK126120/DK/NIDDK NIH HHS/United States
- R01 DK104927/DK/NIDDK NIH HHS/United States
- R00 HG009917/HG/NHGRI NIH HHS/United States
- R01 HG012367/HG/NHGRI NIH HHS/United States
- UM1 HG009435/HG/NHGRI NIH HHS/United States
- R01 HG012872/HG/NHGRI NIH HHS/United States
- UM1 HG009436/HG/NHGRI NIH HHS/United States
- U24 HG009446/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
