

Integrative polygenic risk score improves the prediction accuracy of complex traits and diseases
- PMID: 38508198
- PMCID: PMC11019356
- DOI: 10.1016/j.xgen.2024.100523
Integrative polygenic risk score improves the prediction accuracy of complex traits and diseases
Abstract
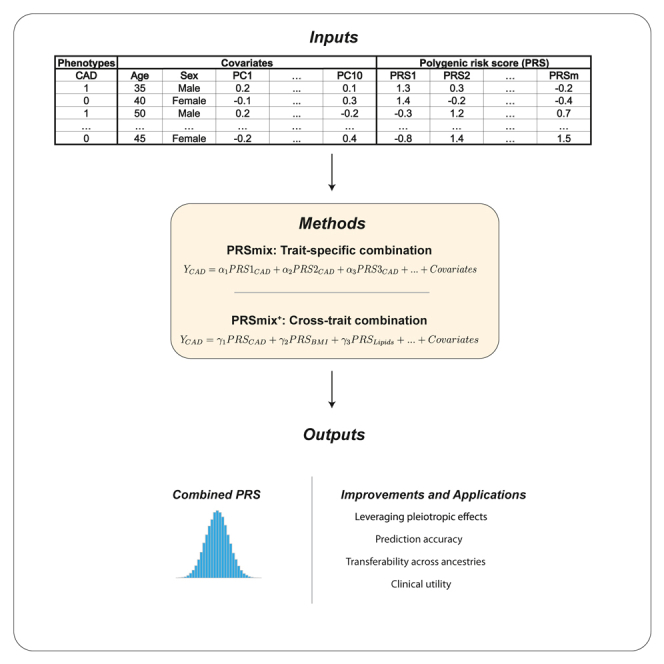
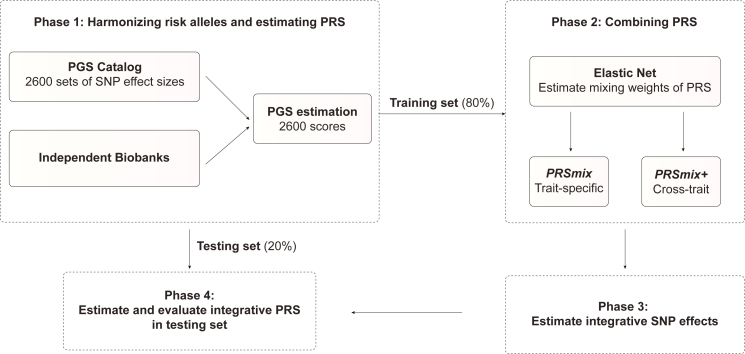
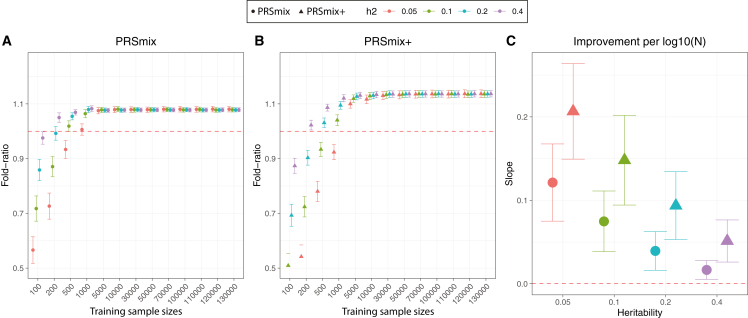
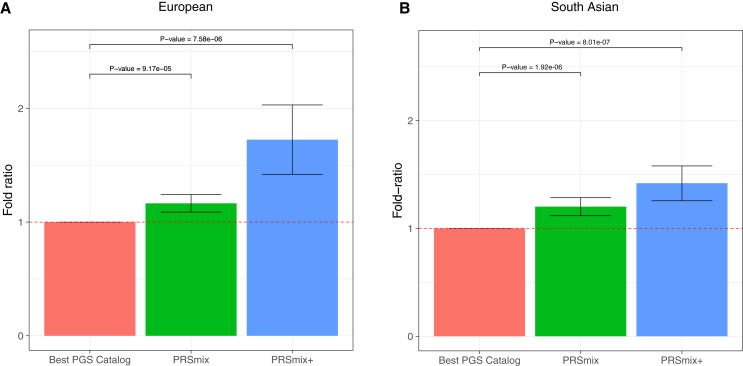
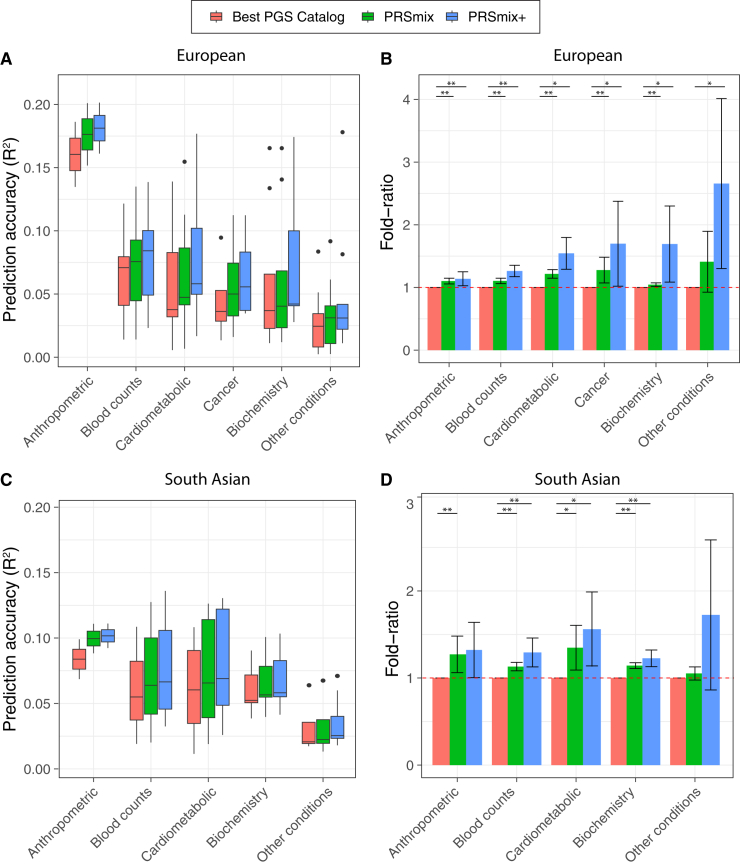
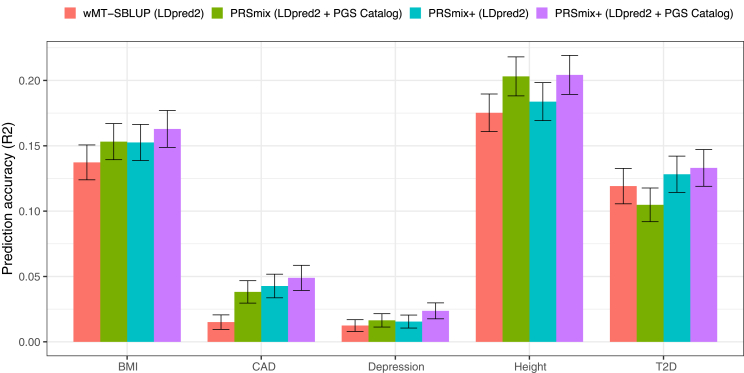
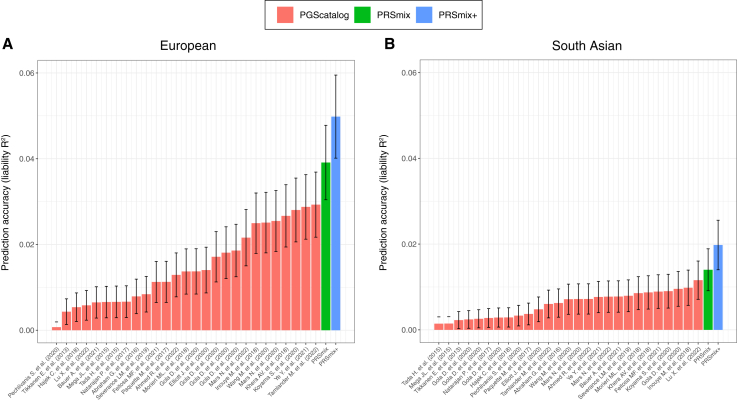
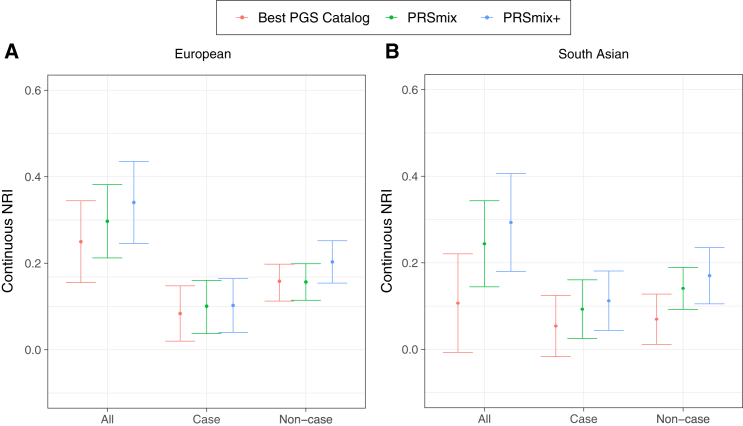
Polygenic risk scores (PRSs) are an emerging tool to predict the clinical phenotypes and outcomes of individuals. We propose PRSmix, a framework that leverages the PRS corpus of a target trait to improve prediction accuracy, and PRSmix+, which incorporates genetically correlated traits to better capture the human genetic architecture for 47 and 32 diseases/traits in European and South Asian ancestries, respectively. PRSmix demonstrated a mean prediction accuracy improvement of 1.20-fold (95% confidence interval [CI], [1.10; 1.3]; p = 9.17 × 10-5) and 1.19-fold (95% CI, [1.11; 1.27]; p = 1.92 × 10-6), and PRSmix+ improved the prediction accuracy by 1.72-fold (95% CI, [1.40; 2.04]; p = 7.58 × 10-6) and 1.42-fold (95% CI, [1.25; 1.59]; p = 8.01 × 10-7) in European and South Asian ancestries, respectively. Compared to the previously cross-trait-combination methods with scores from pre-defined correlated traits, we demonstrated that our method improved prediction accuracy for coronary artery disease up to 3.27-fold (95% CI, [2.1; 4.44]; p value after false discovery rate (FDR) correction = 2.6 × 10-4). Our method provides a comprehensive framework to benchmark and leverage the combined power of PRS for maximal performance in a desired target population.
Keywords: PRS; South Asian; clinical utility; combination; cross ancestry; integrative.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.N. reports grants from Allelica, Amgen, Apple, Boston Scientific, Genentech, and Novartis; is a consultant to Allelica, Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, HeartFlow, Novartis, Genentech, and GV; reports scientific advisory board membership with Esperion Therapeutics, Preciseli, and TenSixteen Bio; is a scientific co-founder of TenSixteen Bio; and reports spousal employment at Vertex Pharmaceuticals, all unrelated to the present work.
Figures
Update of
-
Integrative polygenic risk score improves the prediction accuracy of complex traits and diseases.medRxiv [Preprint]. 2023 Mar 23:2023.02.21.23286110. doi: 10.1101/2023.02.21.23286110. medRxiv. 2023. Update in: Cell Genom. 2024 Apr 10;4(4):100523. doi: 10.1016/j.xgen.2024.100523. PMID: 36865265 Free PMC article. Updated. Preprint.
References
-
- Catalog, P.G.S. PGS Catalog - the Polygenic Score Catalog. http://www.pgscatalog.org/.
-
- Choi S.W., O’Reilly P. SA20 - PRSice 2: POLYGENIC RISK SCORE SOFTWARE (UPDATED) AND ITS APPLICATION TO CROSS-TRAIT ANALYSES. Eur. Neuropsychopharmacol. 2019;29:S832. doi: 10.1016/j.euroneuro.2017.08.092. - DOI
MeSH terms
Grants and funding
- OT2 OD026556/OD/NIH HHS/United States
- U2C OD023196/OD/NIH HHS/United States
- OT2 OD026551/OD/NIH HHS/United States
- OT2 OD026552/OD/NIH HHS/United States
- OT2 OD026549/OD/NIH HHS/United States
- OT2 OD025337/OD/NIH HHS/United States
- R01 HL142711/HL/NHLBI NIH HHS/United States
- OT2 OD025277/OD/NIH HHS/United States
- OT2 OD026550/OD/NIH HHS/United States
- OT2 OD023205/OD/NIH HHS/United States
- OT2 OD025276/OD/NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- OT2 OD026557/OD/NIH HHS/United States
- OT2 OD026554/OD/NIH HHS/United States
- U24 OD023163/OD/NIH HHS/United States
- OT2 OD023206/OD/NIH HHS/United States
- U24 OD023176/OD/NIH HHS/United States
- OT2 OD026548/OD/NIH HHS/United States
- U01 HG011719/HG/NHGRI NIH HHS/United States
- OT2 OD025315/OD/NIH HHS/United States
- U24 OD023121/OD/NIH HHS/United States
- R01 HL127564/HL/NHLBI NIH HHS/United States
- R01 HL151152/HL/NHLBI NIH HHS/United States
- OT2 OD026555/OD/NIH HHS/United States
- K08 HG012221/HG/NHGRI NIH HHS/United States
- OT2 OD026553/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
