

Dynamics of accessible chromatin regions and subgenome dominance in octoploid strawberry
- PMID: 38509076
- PMCID: PMC10954716
- DOI: 10.1038/s41467-024-46861-0
Dynamics of accessible chromatin regions and subgenome dominance in octoploid strawberry
Abstract
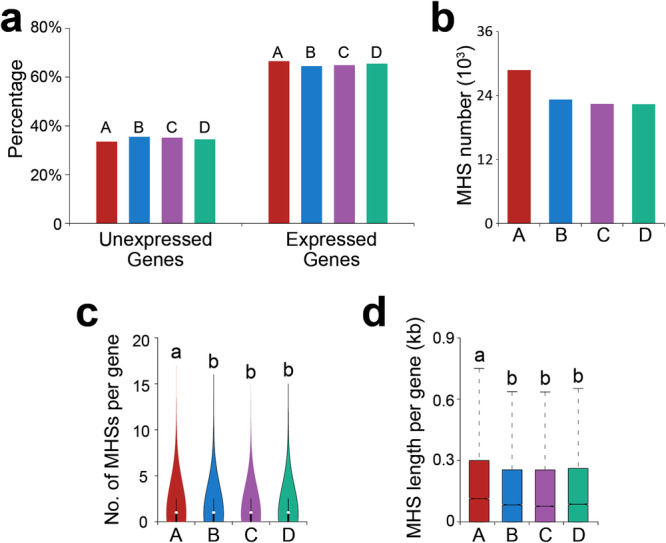
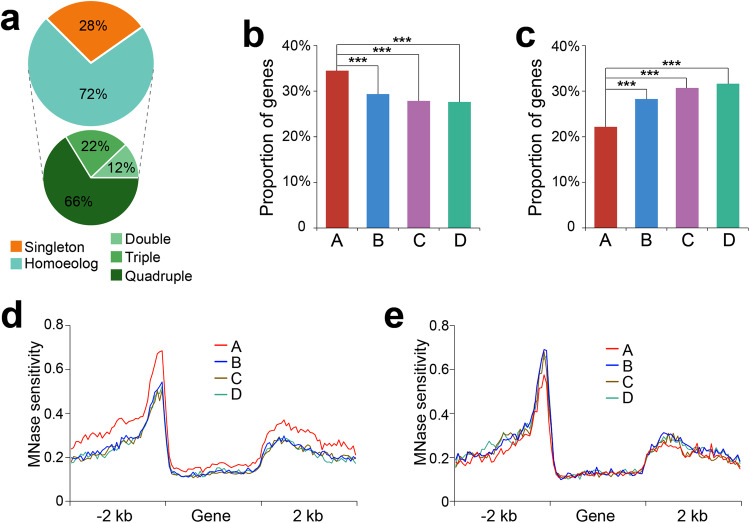
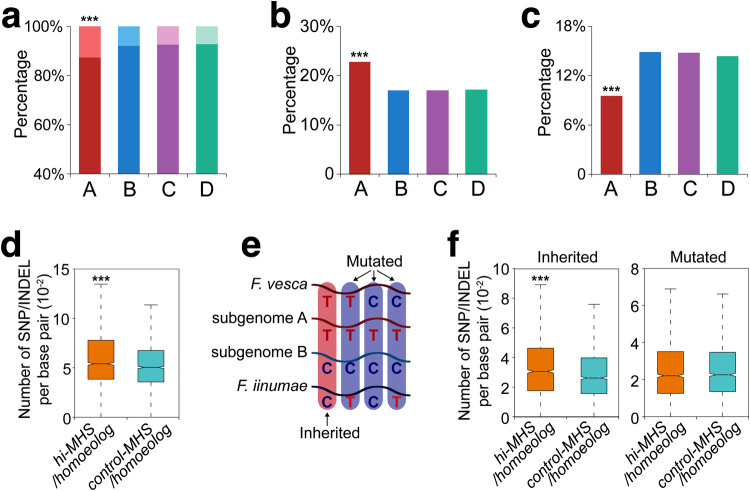
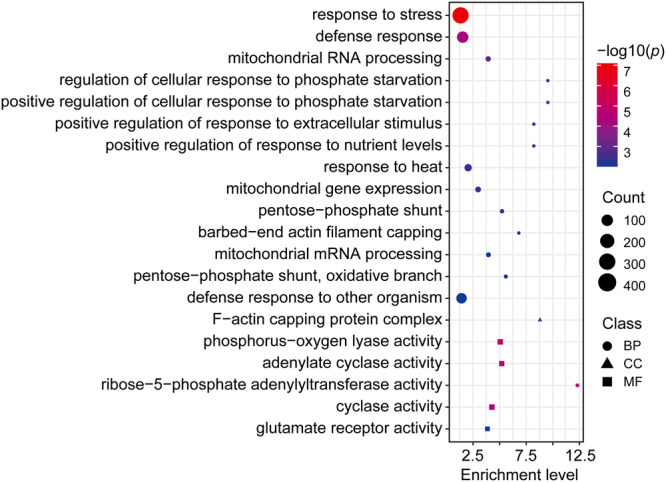
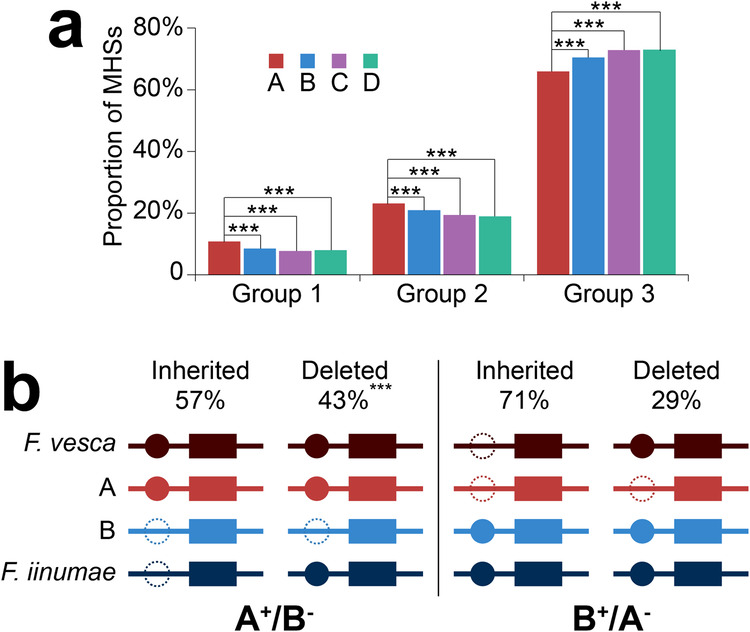
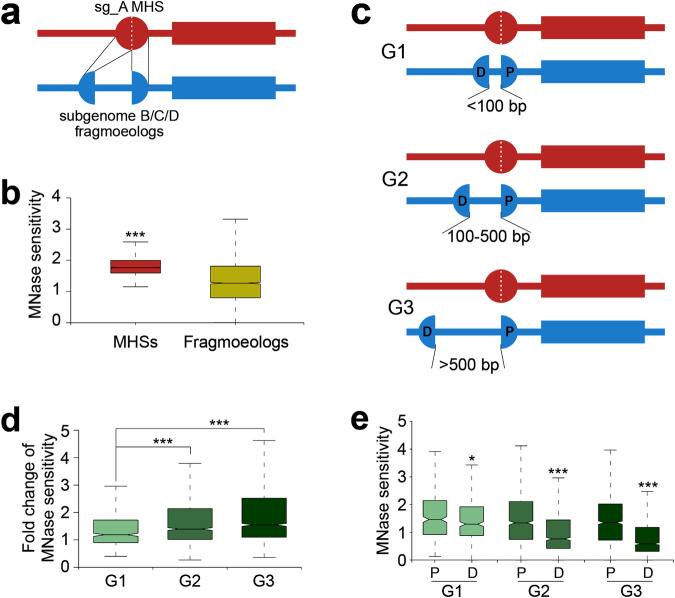
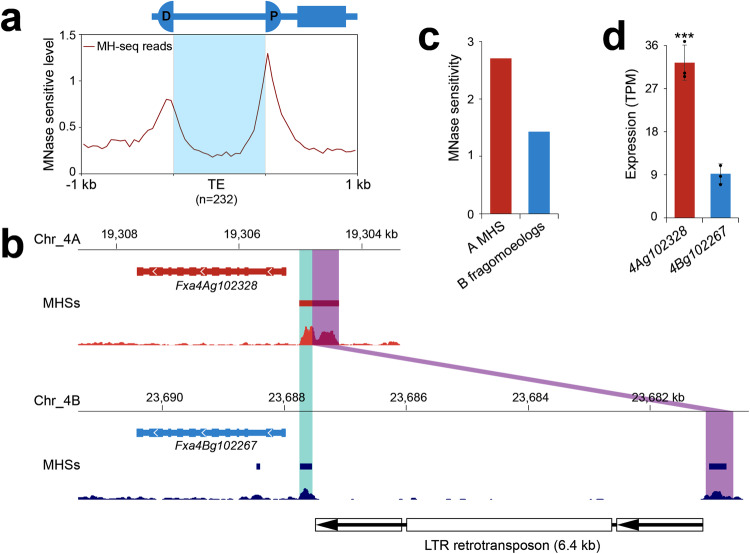
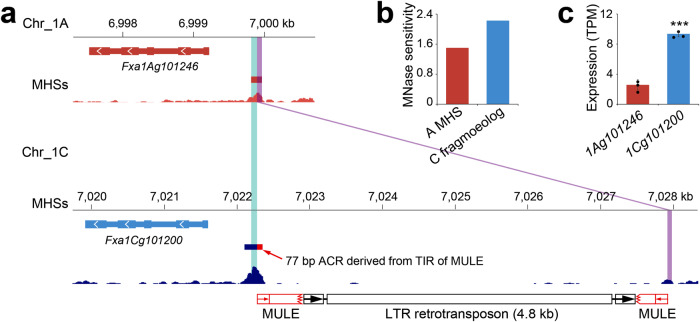
Subgenome dominance has been reported in diverse allopolyploid species, where genes from one subgenome are preferentially retained and are more highly expressed than those from other subgenome(s). However, the molecular mechanisms responsible for subgenome dominance remain poorly understood. Here, we develop genome-wide map of accessible chromatin regions (ACRs) in cultivated strawberry (2n = 8x = 56, with A, B, C, D subgenomes). Each ACR is identified as an MNase hypersensitive site (MHS). We discover that the dominant subgenome A contains a greater number of total MHSs and MHS per gene than the submissive B/C/D subgenomes. Subgenome A suffers fewer losses of MHS-related DNA sequences and fewer MHS fragmentations caused by insertions of transposable elements. We also discover that genes and MHSs related to stress response have been preferentially retained in subgenome A. We conclude that preservation of genes and their cognate ACRs, especially those related to stress responses, play a major role in the establishment of subgenome dominance in octoploid strawberry.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Discrimination of candidate subgenome-specific loci by linkage map construction with an S1 population of octoploid strawberry (Fragaria × ananassa).BMC Genomics. 2017 May 12;18(1):374. doi: 10.1186/s12864-017-3762-y. BMC Genomics. 2017. PMID: 28499415 Free PMC article.
-
Origin and evolution of the octoploid strawberry genome.Nat Genet. 2019 Mar;51(3):541-547. doi: 10.1038/s41588-019-0356-4. Epub 2019 Feb 25. Nat Genet. 2019. PMID: 30804557 Free PMC article.
-
HaploSNP affinities and linkage map positions illuminate subgenome composition in the octoploid, cultivated strawberry (Fragaria×ananassa).Plant Sci. 2016 Jan;242:140-150. doi: 10.1016/j.plantsci.2015.07.004. Epub 2015 Jul 10. Plant Sci. 2016. PMID: 26566832
-
One subgenome to rule them all: underlying mechanisms of subgenome dominance.Curr Opin Plant Biol. 2020 Apr;54:108-113. doi: 10.1016/j.pbi.2020.03.004. Epub 2020 Apr 25. Curr Opin Plant Biol. 2020. PMID: 32344327 Review.
-
The causes and consequences of subgenome dominance in hybrids and recent polyploids.New Phytol. 2018 Oct;220(1):87-93. doi: 10.1111/nph.15256. Epub 2018 Jun 8. New Phytol. 2018. PMID: 29882360 Review.
Cited by
-
Editorial: Interspecific hybridization in plant biology, volume II.Front Plant Sci. 2024 Apr 26;15:1412622. doi: 10.3389/fpls.2024.1412622. eCollection 2024. Front Plant Sci. 2024. PMID: 38736442 Free PMC article. No abstract available.
-
Chromatin Accessibility and Gene Expression Vary Between a New and Evolved Autopolyploid of Arabidopsis arenosa.Mol Biol Evol. 2024 Oct 4;41(10):msae213. doi: 10.1093/molbev/msae213. Mol Biol Evol. 2024. PMID: 39404085 Free PMC article.
-
Natural neopolyploids: a stimulus for novel research.New Phytol. 2025 Apr;246(1):78-93. doi: 10.1111/nph.20437. Epub 2025 Feb 14. New Phytol. 2025. PMID: 39953679 Free PMC article. Review.
-
A near-complete genome assembly of Fragaria iinumae.BMC Genomics. 2025 Mar 14;26(1):253. doi: 10.1186/s12864-025-11440-0. BMC Genomics. 2025. PMID: 40087556 Free PMC article.
References
-
- Ohno, S. Evolution by Gene Duplication. (Springer Berlin, 1970).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
