

doi: 10.1038/s41592-024-02229-2.
Epub 2024 Mar 20.
SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms
Affiliations
- PMID: 38509328
- PMCID: PMC11093726
- DOI: 10.1038/s41592-024-02229-2
Item in Clipboard
SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms
Nat Methods.
2024 May.
Abstract
SQANTI3 is a tool designed for the quality control, curation and annotation of long-read transcript models obtained with third-generation sequencing technologies. Leveraging its annotation framework, SQANTI3 calculates quality descriptors of transcript models, junctions and transcript ends. With this information, potential artifacts can be identified and replaced with reliable sequences. Furthermore, the integrated functional annotation feature enables subsequent functional iso-transcriptomics analyses.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests
Figures
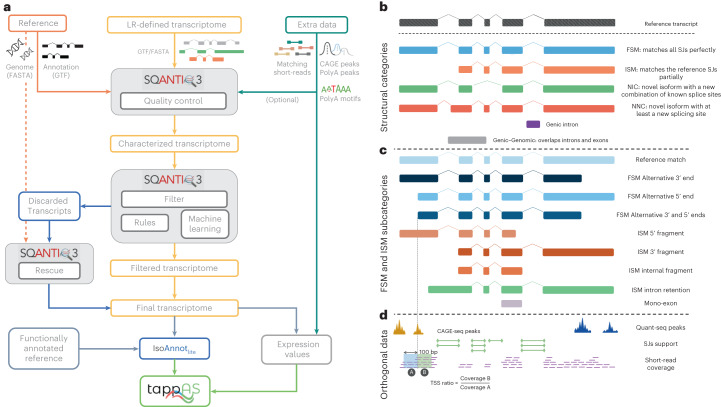
a, SQANTI3 workflow. b, Main SQANTI structural categories for transcript models of known genes. c, SQANTI3 subcategories for FSM and ISM transcripts. d, Orthogonal data features processed by SQANTI3 QC. LR, long read; SJ, splice junction.
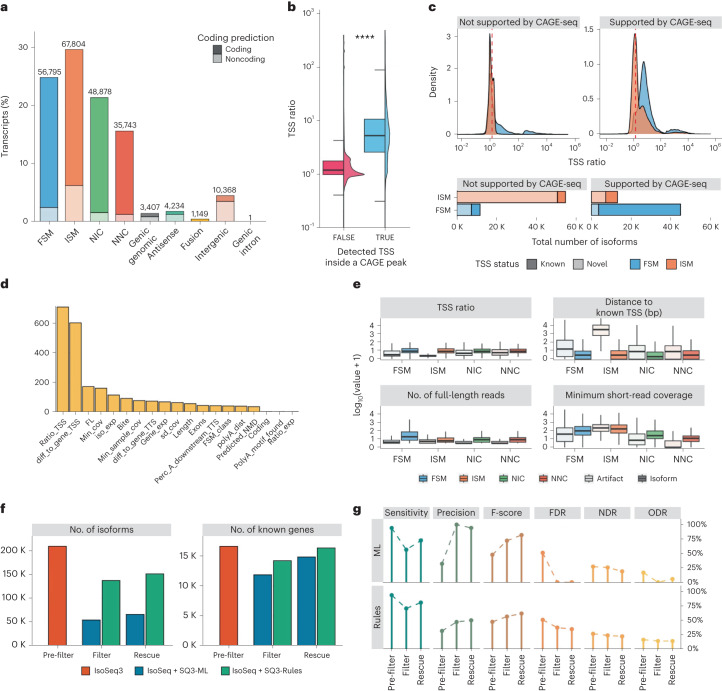
a, Distribution of transcript models by SQANTI3 structural categories in the WTC11 IsoSeq3-defined transcriptome. b, Differences in TSS ratio between TSS supported and not supported by CAGE-seq data (****P = 2 × 10−16, two-sided Wilcoxon test). c, Uneven support of CAGE-seq data for FSM and ISM transcript models, with supported TSS usually having a TSS ratio > 1.5 (red dashed line), particularly if they are also known TSS. d, Variable importance of SQANTI3 descriptors in the machine learning (ML) filter for different input scenarios, obtained after training the random forest model on different true-positive (TP) sets. e, Distribution of values for the top machine learning filter variables (ranked by random forest classifier importance) for isoforms and artifacts across the FSM (35,446 isoforms, 21,079 artifacts), ISM (2,573 isoforms, 65,114 artifacts), NIC (15,504 isoforms, 33,367 artifacts) and NNC (1,556 isoforms, 34,182 artifacts) structural categories. f, Variation in the number of genes and transcripts after filter and rescue using sample-specific orthogonal data with rules and machine learning approaches. g, Performance metrics according to SIRV detection at each step of the SQANTI3 pipeline using rules and machine learning approaches. FDR, false discovery rate; NDR, novel detection rate; ODR, over-annotation detection rate For all boxplots in this figure, the middle line represents the median, the ends of the box represent the 25th (quartile 1) and 75th (quartile 3) percentiles, and the whiskers represent the minimum (quartile 1 minus 1.5-fold the interquartile range (IQR)) and the maximum (quartile 3 plus 1.5-fold the IQR). The half-violin plots show the density distribution of values.

1) If an FSM-supported reference transcript is lost during the filtering, the version of the reference is automatically rescued. 2) The rest of the LR-defined transcript models filtered out (rescue candidates) are mapped against the reference transcriptome combined with the accepted LR-defined isoforms (rescue targets), allowing several hits per candidate. 3) Reference transcriptome was previously evaluated and filtered with the same data and criteria as the LR-defined transcripts. 4) Rescue is completed by evaluating targets. They need to pass the filtering and not increase the redundancy, meaning that if the target is an LR-defined transcript present or it is a reference transcript already represented as an FSM in the filtered transcriptome, these targets will not be added to the final annotation. LR: Long-read, ML: Machine Learning, FSM: Full-Splice-Match, ISM: Incomplete-Splice-Match, NIC: Novel-In-Catalog, NNC: Novel-Not-In-Catalog.

Number of TSS identified using the TSS ratio (threshold=1.5) based on matching short-reads RNA-seq data, sample-specific CAGE-seq data and the refTSS database. TSS: Transcript Starting Site.

Number of TTS identified using sample-specific Quant-seq data, presence of polyA motif and the PolyASite database. WTC11 PacBio lrRNA-seq data. TTS: Transcript Termination Site.

Data are stratified by SQANTI3 structural category and separated according to the existing Quant-seq data support. WTC11 PacBio lrRNA-seq data.

Data are broken-down by SQANTI3 structural category and separated depending on whether transcript models were flagged as potential intrapriming artifact. Boxes indicate median (middle line), 25th (Q1) and 75th (Q3) percentiles (box hinges); whiskers represent min = Q1 - 1.5 ⋅ Interquartile Range (IQR) and max = Q3 + 1.5 ⋅ IQR; dots constitute outliers. FSM: Full-Splice-Match. ISM: Incomplete-Splice-Match, NIC: Novel-In-Catalog, NNC: Novel-Not-In-Catalog.

Rescue candidates are shown in the y-axis, stratified by structural category. Candidates correspond to transcripts discarded by the ML filter, that is artifacts. Rescue targets are shown in the x-axis, spread across structural categories and including reference transcriptome hits. Targets correspond to transcripts mapped by artifacts during the rescue process. In this mapping process, each candidate can map to multiple targets, which are similar to the candidate in sequence and exon structure. Heatmap color therefore corresponds to the number of hits (log10) involving each possible pair of structural categories, indicating the amount of structural similarity among categories detected during rescue. Within the tiles, the total number of candidate target pairs is shown, including the mean number of hits per candidate for each category pair between parentheses. FSM candidates only match reference targets, since they are only considered for automatic rescue. WTC11 PacBio lrRNA-seq data. FSM: Full-Splice-Match, ISM: Incomplete-Splice-Match, NIC: Novel-In-Catalog, NNC: Novel-Not-In-Catalog.

a, Distribution of expression values (TPM) of known transcripts detected as Full-Splice-Match or Incomplete-Splice-Match. b, TRIFID scores of known transcripts identified in each filtering and rescue scenario. Filtered transcripts (orange) did not pass the corresponding filter and were not eventually rescued. Transcripts filtered but recovered by introducing an isoform from the reference (dark blue) represent the rescue strategy’s fundamental purpose. In exceptional cases, transcripts models not initially detected were included in the final transcriptome (yellow) via rescue.
Update of
-
SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms.bioRxiv [Preprint]. 2023 Jun 3:2023.05.17.541248. doi: 10.1101/2023.05.17.541248. bioRxiv. 2023. Update in: Nat Methods. 2024 May;21(5):793-797. doi: 10.1038/s41592-024-02229-2. PMID: 37398077 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
